
Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome)


Abstract
ZEB2 and TCF4 are transcription factors (TFs) whose locations in embryos overlap in many sites and developmental phases, including in the forebrain and its cortical neurons. De novo mutations cause the phenotypically overlapping, haploinsufficient Mowat-Wilson (MOWS, in the ZEB2 gene) and Pitt-Hopkins (PTHS, in TCF4) syndromes, which currently cannot be cured. Mutant alleles have been mapped and defects documented (also in brain function) in MOWS and PTHS patients. Appropriately designed mouse models and cells derived from these, as well as cellular models including cultured pluripotent cells, enable investigating the genetic and molecular mechanisms underlying the developmental deficiencies that manifest after birth in the nervous systems and their multiple cell types, as well as those of organs other than the brain, in MOWS and PTHS. Biochemical analyses of cell type-specific transcriptomic changes in these perturbation models as compared to control cells, the identification of the intact-factor dependent and direct target genes, and of partner proteins including chromatin modulators, are revealing complex and multiple modes of action that eventually will explain target gene selectivity for these TFs. Both TFs have also been found to operate in acute and chronic diseases and cell-based repair processes after tissue or organ injury. In addition, the defective function also arises from their aberrant gene expression, which will require a deeper investigation of how the transcription of these TF genes is regulated. Furthermore, these two factors genetically and biochemically interact. This review combines the essentials and recent progress for both TFs for the first time, with a focus on MOWS and PTHS.
Keywords
INTRODUCTION
Many causal genes have been identified, and subsequently, de novo mutations in patient cohorts mapped in syndromes that manifest before, early, or somewhat later after birth with neurodevelopmental defects such as microcephaly, intellectual disability, seizures, and epilepsy. In many of the monogenic neurodevelopmental syndromes, it is striking that the mutation occurs in a gene encoding a brain development transcription factor (TF) or a subunit of chromatin remodeling complexes (e.g., SOX11[1]; a number of zinc finger TFs[2]; NR2F1[3]; chromodomain DNA-binding helicases CHD3[4] and CHD8[5]; and subunits of the ATP-dependent Mi-2/nucleosome remodeling and deacetylase (NuRD) co-repressor complex[6]; see also the review in Ref.[7]). Here, we discuss two members of different TF-families that have joined this stage: ZEB2 (originally named SIP1, on human chr2q22.3) of the ZEB family and TCF4 of the basic helix-loop-helix (bHLH) family (originally named E2-2, on human chr18q21.2, and not to be confused with T-cell factor-4, renamed TCF7L2).
Heterozygous mutations in ZEB2 or TCF4 cause Mowat-Wilson syndrome (MOWS, OMIM #235730) or Pitt-Hopkins syndrome (PTHS, OMIM #610954), respectively. MOWS and PTHS have strong phenotypic clinical overlap, which warrants interest in co-monitoring the recent progress in ZEB2 and TCF4 research. Indeed, one starts to compare in detail their multiple functions and explore their possible mechanistic overlap and genetic interaction. As a result, knowledge is emerging that ZEB2 and TCF4 may also be modifier genes of one another, as well as act as modifiers in other neurodevelopmental syndromes and psychiatric disorders (for ZEB2, see Ref.[8]; for TCF4, see Ref.[9]). In addition, analyses of ZEB2- or TCF4-perturbed models range from neuronal and glial cells to other cell types outside of the nervous systems, i.e., the hematopoietic and immune system, cardiac malformation and malfunction, digestive tract pathology, or even cancer. These features may incite clinical geneticists to re-consider the broad defects of MOWS and PTHS patients in adulthood and to follow up on these patients longitudinally from these new angles as well.
EXPERIMENTAL APPROACHES PUT IN CONTEXT
In addition to the initial mapping of ZEB2 and TCF4 expression sites in the developing brain, the studies of upstream regulations and downstream effects of each causal gene/protein in neurons and glial cells, and/or the complex(es) these TFs are incorporated in, have become of highest relevance. Such studies are contributing strongly to the emerging principle of their molecular and phenotypic convergence, which is anticipated with other TFs or co-factors to apply more widely to other neurodevelopmental syndromes or neuropsychiatric disorders[10].
The ongoing studies identify factor-dependent and direct target genes and/or additional protein partners in brain and neural stem/progenitor cells (NSCs and NPCs) during cell diversity creation in the developing brain. Recently, studies have also started to address brain expansion in evolution (in NSCs and NPCs, e.g., for human CHD8[11], mouse Sox2 and Chd7[12]; multiple regulatory proteins[13]; Tcf4, Olig2, and Npas3[14]; and FOXP[15]). They are often complemented with epigenetics and chromatin conformation change studies, which contribute to a better understanding of the precise regulation of their gene expression (see reviews[16-18]; e.g., studies of TRRAP, a phosphoinositide 3-kinase-related kinase present in histone acetyltransferase (HAT) complexes[19]). They also include studies of post-translational modifications of the factors, often leading to a better understanding of their activities and/or controlled incorporation in - or removal from - complexes. Such global biochemical insight eventually helps to better delineate diseases or syndromes. This is mainly achieved by demonstrating and explaining the selective transcriptional control of the factor-bound genes, as well as the impact of mutation or changed dosage of the factor on (each of) the factor functions. This also applies to ZEB2/MOWS and TCF4/PTHS.
Often, the protein-coding sequence of interest is genetically perturbed in mice, using many different conditional knockouts (cKOs), which for the brain is facilitated by the large panel of brain cell-type specific Cre-recombinase strains that are available. However, the increased mapping of missense mutations in particular, and single nucleotide polymorphisms (SNPs) and regulatory elements (REs; e.g., enhancers) of the causal gene(s) in patients with neurodevelopmental syndromes, now allows developing faster, more elegant, and more diverse novel cell-based models rather than entire embryos or animals. Furthermore, such embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and NSCs/NPCs derived from these and grown in cell culture can be submitted to differentiation protocols. For early neural differentiation and subtype neuron specification (and still less for terminal maturation of neurons), these protocols work efficiently in 2D and 3D cultures. These include dorsal or ventral types of forebrain cells in spheroids (including fused spheroids) and cerebral organoids[20-28]. Moreover, prior to differentiation, these cells are amenable to genetic perturbation (e.g., by CRISPR-Cas). Multi-omics approaches can be applied during the entire process (for CHD8[29]), even at a single-cell level, to address cell differentiation as well as cell heterogeneity and consider interesting questions about cellular plasticity. Perturbations [KOs, but also RNAi-based knockdown (KD)] also significantly made the research move to less reductionist and, therefore, more systems biology studies. This allowed identifying and modeling the gene/protein regulatory networks (GRNs and PRNs), not only at one or more stages but also during all cell state transitions[30,31], and again at both cell population and single-cell levels.
Here, we illustrate that ZEB2 and TCF4 are excellent TFs to join this experimental stage-as we propose, preferably together-offering exciting insights in healthy neurodevelopment as well as in the respective syndrome they cause.
MOWAT-WILSON AND PITT-HOPKINS SYNDROMES: HISTORY AND MUTATION SPECTRUM
Mowat-Wilson syndrome
Soon after its discovery as one of the first interacting TFs for Smad proteins (major intracellular effector proteins in transforming growth factor type β/bone morphogenetic protein (TGFβ/BMP) family signaling), hence its name Smad-interacting protein-1 (SIP1, renamed ZEB2)[32], three teams of clinical geneticists independently identified de novo mutations in ZEB2 in patients with severe ID, typical facial dimorphism, and Hirschsprung disease (HSCR)[33-35] as the cause of the well-defined, monogenic, and rare Mowat-Wilson syndrome (MOWS)[36-38]. Mutant ZEB2 alleles have been determined for about 350 patients who display a great variety of clinical features[39-44]. Typical in MOWS patients are delays in developmental milestones and motoric development, and anomalies in the eyes and teeth. Other features are typical craniofacial malformation, sensorineural deafness, and HSCR, which originate from defects in the ZEB2-positive (+) cells of the embryonic neural crest cell lineage. Excellent clinical reports have been published on various malformations in the central nervous system (CNS) of MOWS patients over a broad age range, including anomalies of the corpus callosum and/or hippocampus, which can be seen by neuroimaging. These reports have also started to document the longer-term evolution of electro-clinical defects such as focal seizures (by video-electroencephalograph monitoring, video-EEG)[44-46].
The majority (~93%) of severe MOWS cases are caused by large deletions, nonsense mutations, or short deletions/insertions causing a frameshift leading to a premature stop codon. All of these mapped mutations thus far affect the protein-coding sequences and create haploinsufficiency. Rarer (~5%) are frameshift mutations and in-frame deletions affecting the C-terminal part of ZEB2 only. This raises the possibility that, depending on the location of such mutation, the C-terminally truncated protein is still produced and not subject to nonsense-mediated mRNA decay[43,44,47]. Up to date, only few missense ZEB2 mutations (~1.5%) have been detected[48,49]. These two smaller groups of ZEB2/MOWS-confirmed patients, in general, represent the milder and mildest cases, respectively.
The broad spectrum and relative numbers of severe, milder, and mildest cases make it difficult to firmly conclude on genotype-phenotype correlation. Normally, that conclusion would be based on larger panels of often subtly mutated and, therefore, an aberrant function of one or more of the (hitherto known) affected domains within the ZEB2 protein. For example, two mild MOWS cases have mutations in the NuRD interaction motif (NIM, see below), only causing a loss of interaction of this mutant ZEB2 with the NuRD co-repressor complex, and leaving the rest of ZEB2 intact. However, this has already detectable consequences at the level of ZEB2-dependent and direct target genes in the cellular systems where these mutant proteins were tested[50,51].
To the list of mapped MOWS mutations that affect the protein-coding exons of ZEB2 (in patients now grouped as Class I), variation/mutation of non-protein-coding sequences of the ZEB2 locus have also begun to be added by the clinical geneticists and mapped exclusively to untranslated exons or (distal) REs (the patients being referred to as Class II). Co-operative enhancers active in NPCs were recently identified[52] via mapping of chromatin conformational changes in the large genomic region encompassing the human ZEB2 and mouse Zeb2 locus (both on chr2q). These neural enhancers are located about 500 kilobases (kb) away from the transcriptional start site (TSS) of ZEB2/Zeb2, in a ~3.5 megabase (Mb)-long gene desert upstream of the gene[52]. These add to recently identified, different Zeb2 enhancers in fetal and adult hematopoiesis in mice[53]. Such findings may make clinical geneticists broaden the current characterization of sequenced MOWS alleles, in particular in mild but clinically convincing MOWS patients, wherein no mutation can be found by exon sequencing of ZEB2. In addition, such new non-protein-coding mutations will inspire establishing new cellular or animal models and documenting deviating ZEB2-dependent transcriptomes and ZEB2-containing GRNs in these.
Pitt-Hopkins syndrome
After the first report[54] in 1978, it has taken quite some time to delineate the few similar syndromic cases and eventually claim the new syndrome Pitt-Hopkins syndrome (PTHS), based on three defects: (i) brain-related defects (including ID, often coming to attention in the first year of life); (ii) craniofacial malformation; and (iii) episodes of hyperbreathing that start in infancy, as well as breath holding and cyanosis. Several teams confirmed that de novo TCF4 haploinsufficiency causes PTHS; thus, not the phenotypically overlapping Angelman (OMIM #105830), Rett (OMIM #312750), alpha-thalassemia/mental retardation syndrome, and X-linked (ATR-X; OMIM #301040) syndromes[55-57], as well as not MOWS, identified only around the year 2000[33-35]. The situation is somewhat more complicated because of the existence of Pitt-Hopkins-like syndromes 1 and 2 (PTHLS1/2, OMIM #610042 and #614325) caused by autosomal recessive variants in CNTNAP2 and NRXN1, respectively, with patients who in general have milder delays in motor development.
The original identification of the PTHS locus benefited from large (> 1 Mb) deletions on chr18q in patients, enabling the use of SNP arrays for molecular karyotyping, and was further facilitated by sequencing of many coding exons (and intronic flanking regions) of TCF4, one of the candidate genes in this deletion region. Subsequent selection of extra patients with at least two out of the three defects (see (i)-(iii) above), and exclusion of the other similar syndromes, then confirmed the 20-exon (ex) (with ex1 and ex20 considered non-protein-coding at that time) and at least 360 kb-long TCF4 gene as the cause of PTHS. Increased numbers of cases thereafter allowed clinically delineating PTHS, aided by the use of two different clinical tests. These diverged due to the difference in weight given to some of the applied criteria (such as facial features, ID, and gait), and the interpretation of the test score also varied with age[58,59]. PTHS often includes severe psychomotor delay, seizures and epilepsy, and stereotypical and repetitive movements. The same holds for microcephaly, but minor brain abnormalities are revealed by imaging (dysplasia of cerebellum and corpus callosum, small hippocampus, and hyperintensity of temporal lobe white matter). Additional defects are eye defects (early-onset myopia and strabismus), wider spacing of teeth, anomalies of fingers and toes, chronic constipation, and intestinal malrotation[58,60]. Thus, many of these defects overlap with those in MOWS[8,9,44-46,58,59].
The wide phenotypic spectrum and possible clinical inclusion bias, in general, have prompted a number of teams to join forces and apply ID-targeted high-throughput sequencing (HTS) of hundreds of genes in ~900 patients with moderate to profound ID, but without the firm mapping of their syndrome[61]. Doing so, as part of this study, TCF4 variants were identified in eight of these patients and could be added to two other ones from a previous ID-HTS study. These ten patients were then evaluated via both aforementioned PTHS clinical tests, and five of them scored convincingly positive. These same studies revealed that the TCF4 mutation rate is 0.7% in individuals with undiagnosed ID, but also that mutations that cause ID are poorly suggestive of PTHS (and therefore had sometimes been referred to as nonspecific ID). Additional large patient cohorts (~280 patients) assembled from 58 publications with both clinical and sequence data yielded another 25 case reports that were re-investigated[62].
Altogether, this well-coordinated and vast screening made clinicians aware that in PTHS, more precise diagnostic criteria as well as detailed phenotyping were needed. A survey of hundreds of children with PTHS allowed an estimation of missense (~15% of cases), nonsense (15%), and splice-site (10%) mutations. In addition, small insertions/deletions causing a frameshift (30%), larger deletions, and even translocations involving a part of or the entire TCF4 (see Ref.[9], and references therein) have been detected. Most missense TCF4 mutations, which affect all isoforms, map to ex19 (encoding the bHLH domain, see below), although some are located in ex15 (middle part of AD2) or ex18 (Rep or RD), and a few cases of C-terminally elongated TCF4 in mild PTHS have been described.
ZEB2 AND TCF4: PROTEINS, AND GENES AND THEIR REGULATION
ZEB2 protein/gene structure
Mouse Zeb2 [1215 amino acids (aa); human ZEB2 (1214 aa; ~140 kDa) was after mouse Zeb1, the second identified member of the small family of vertebrate ZEBs. ZEB1 and ZEB2 are structurally highly similar (for a schematic representation of ZEB2, see Figure 1A]: they have a NIM close to their N-terminus and, more downstream, a homeodomain-like domain (HD, but which does not bind to DNA) and a C-terminal binding protein (CtBP)-interacting domain (CID) with four interaction motifs for (the non-DNA-binding) CtBP1/2 co-repressors. ZEB proteins contain two separated clusters of (between ZEB1 and ZEB2) strongly conserved zinc fingers. The N-terminal (NZF, with four fingers) and C-terminal cluster (CZF, with three fingers) enable ZEB proteins to bind with their two hands (and two zinc fingers in each) to DNA. Binding occurs to two separated CACCT sequences[63,64], often also found as CACCTG known as E2-boxes, and in few cases also presenting as CACANNTG[8,52] (CANNTG being an E-box). This could make ZEB proteins putative competitors of bHLH TFs for the same E-type boxes, including for the TCF4-containing heterodimers that bind to the CANNTG E-box, with a preference for C or G in the N positions[65]. In the cell nucleus, ZEB2 (not ZEB1) detectably binds to receptor-activated phospho(p)-Smad proteins that act downstream of liganded TGFβ/Nodal/BMP family receptors. In ZEB2’s short (defined as 14 aa-long) Smad-binding domain, a tandem repeat of QxVx is crucial for binding to all p-Smads[32,66].
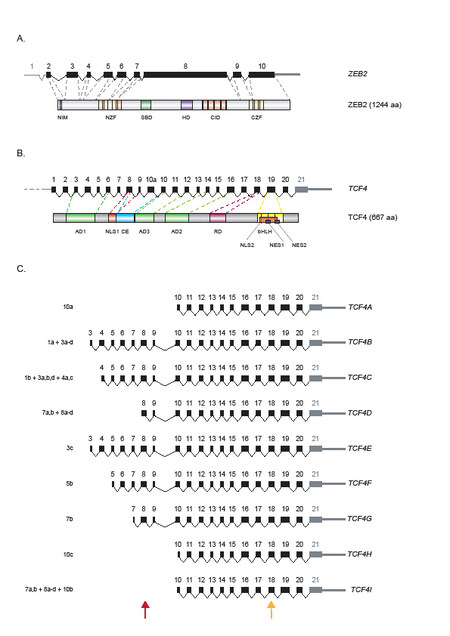
Figure 1. Schematic overview of ZEB2 and TCF4. (A) Representation of (human) ZEB2 gene exons/introns (top) and ZEB2 protein structure (bottom). The largest ex8 encodes for the largest protein segment (in mouse, this corresponds to ex7). NIM: NuRD-interacting motif; NZF and CZF: N- and C-terminal zinc finger clusters; SBD: Smad-binding domain; HD: homeodomain-like domain; CID: CtBP-interacting domain (see Ref.[8]). (B) TCF4 gene protein-coding exons (20 in total), the thereafter identified 3’ untranslated exon (ex21) and introns (topl), and TCF4 protein structure (bottom). For reasons of huge complexity, the numerous other identified 5’-non-coding exons and various TSSs (indicated minimally as …) are not included here. AD: Transcription activation domain; NLS: nuclear localization signal; CE: conserved element; RD: repression domain (sometimes indicated as Rep); bHLH: basic helix-loop-helix DNA-binding domain; NE: nuclear export signal. (C) Alternative TCF4 transcripts as determined for brain. Untranslated regions are not included in this drawing, so only the used protein-coding exons are drawn (in black), together with the one 3’-untranslated ex21 (in grey). On the left, the number/letter combinations indicate that these transcripts also contain brain-specific exons (of which the numbers are given) that are still incorporated in the transcripts, but not coding for protein in these cases, while the letters indicate different mapped splice sites. Names of the transcripts are indicated on the right. The brown arrowhead at the bottom of the panel indicates the region of alternative splicing resulting in a full-length or a so-called Δ variant. The orange arrow at the bottom indicates the location of the splicing causing the generation of the so-called - and + isoforms. The images in (B, C) are based on the original report[67] and inspired by reviews[9,113].
In most assays, ZEBs act as transcriptional repressors of tested target genes or transfected promoter-reporter combinations, but, by interacting with histone acetyltransferase (HAT) containing P300 or the related CREB-binding protein (CBP), they can also activate (other) sets of genes (see review Ref.[52], and references therein). The mouse Zeb2 locus contains upstream of the first translated exon altogether nine untranslated exons (named U1-U9), which via alternative splicing, are joined to the protein-encoding parts of the Zeb2 transcript, however, without resulting in Zeb2 protein variants. The longest protein segment encoding exon in mice is ex7, corresponding to ex8 in humans [Figure 1A].
TCF4 protein/gene structure
The structure of the TCF4 gene is very complex: through the subsequent mapping of more alternative 5’-exons [20 in total, not shown in Figure 1B], the demonstration of 20 coding exons and the confirmation of one previously unreported 3’-terminal non-protein-coding exon [Figure 1B], the total number of exons in TCF4 has now risen to 41[67] (for the equal complexity of mouse Tcf4 alternative transcripts, see Ref.[68]; for subsequent reviews, see Refs.[9,69]; also note that Ensembl lists 48 transcripts) and the size of the human gene accordingly to > 400 kb. Together with alternative TSSs upstream of seven exons in total, this TCF4 human gene structure is proposed to yield at least 18 protein isoforms (TCF4-A to TCF4-R), which differ at their N-terminus; at least 13 of these are present in the brain. The numbers of different mRNA transcripts are even higher due to alternative splicing within the protein-coding exons [indicated in Figure 1C]. Alternative 5’-exons are used in TCF4, also resulting in transcripts that only include ex7-20. To our knowledge, it remains to be established firmly whether human iPSCs or differentiated organoids (or other ex vivo models) recapitulate this huge diversity in TCF4 transcripts, its heterogeneity between cell types, and whether this is also the case in the mouse. This mission announces difficulty, even when using single-cell RNA-seq, for full-length sequencing of transcripts combined with the highest depth of sequencing will be needed.
The first suggestion of the position of the mutation in TCF4 being relevant to part of the PTHS phenotype, in particular, those aspects being linked to mild non-syndromic ID rather than full PTHS, were mutations in the 5’ region of the gene and cassette exons and regions not affecting the important functional domains of TCF4[70]. Subsequent mapping of TCF4 mutations in PTHS did suggest genotype-phenotype correlation: those within ex1-4 and ex4-6 would cause mild ID, within ex7-8 severe PTHS albeit without overt facial malformation, and those within ex9-19 typical PTHS[71]. These have been suggested to correlate with preservation of the intact nuclear localization signal (NLS) and bHLH domains, and with mutation of the NLS and minimally the bHLH domain (encoded by ex18), respectively. Although a genotype-phenotype paradigm has been proposed based on these observations, this has remained rather unclear to molecular biologists. They likely expected extensive documentation of the function of the mutant TCF4 proteins, their heterodimerization with the Class II, cell-type specific bHLH TFs, and their stability vs. degradation. However, significant attempts at addressing their subnuclear localization and dimerization, including in quantitative terms, and activity on target promoters such as NRXN1β and CNTNAP2 promoters have been carried out. These assays indeed pointed out the multiple effects of PTHS missense mutations on TCF4 global function[72].
HLH heterodimers composed of Class I (seemingly “ubiquitous”, so present in nearly all cell types) and II (clearly cell-type specific) TFs bind to E-boxes in promoters and enhancers (see review Ref.[73]). Their affinity to E-boxes is co-determined by co-factors, as well as by Ca2+-dependent proteins such as calmodulin that interact with the basic (b) stretch in the (b)HLH domain. In addition, flanking DNA sequences influence the binding affinity of these TFs. Indeed, a subset of TCF4-bound sites has the expanded consensus binding specificity 5’-C(A/G)-CANNTG-3’, sometimes with an added downstream outer C(A/G). When present as CpG, both possible additions can, in the mammalian brain, be variably methylated and have positive as well as negative effects on TCF4-binding[74].
Some consequences and extra complexity
Interestingly, the 5’-C(A/G)-CANNTG-3’ motif has also been identified as a binding site for ZEB1 and, due to the high conservation of DNA-binding zinc fingers between ZEBs, been accepted for both ZEB1 and ZEB2[8,63]. For such motif, the influence of methylation (of a 5’-CG) for binding of ZEBs has not been documented yet. In reverse, it is tantalizing that, via the bHLH domain, TCF4 may be involved in sensing cytosine modification, and that this might also be so for ZEB proteins. Similar to ZEB proteins, TCF4 recruits HATs such as P300[75-77]. Additionally, in mouse NPCs, Tcf4 binds Mediator and thus co-defines super-enhancers that maintain cell identity[78].
TCF4 is expressed in many cell types/organs, which classified TCF4 readily as a “ubiquitous” bHLH factor, despite the subsequent identification of cell- and/or tissue-specific presence of the many isoforms [for a schematic representation of TCF4, see Figure 1B]. Furthermore, the aforementioned complexity of its transcripts means that different TCF4 isoforms [for a selection, see Figure 1C; for recent update, see Ref.[9]] contain or do not contain domains such as AD1, NLS1, and CE (then referred to as the Δ isoforms), or that the longer isoforms are the only ones that have a complete AD1 [Figure 1B]. The same holds for alternative incorporation or exclusion of an RSRS-tetrapeptide sequence (named as + are - isoforms, respectively) due to two alternative donor splice sites in ex18. In any case, all TCF4 transcripts have ex10-20, hence encompass AD3 (ex10-12), AD2 (ex14-16), Rep/RD (ex18), and HLH, NLS2, and NES1/2 (ex19).
All this makes the precise mapping of spatial expression and the estimation of the levels of each of the various TCF4 transcripts (and Δ and +/- isoforms) very complex and technically very difficult. This is also the case in the brain, but in general, the majority of transcripts are expressed here [these are specifically shown in Figure 1C], except for those encoding protein isoforms TCF4-J to -N, as these are only present in the testes.
ZEB2 and TCF4 gene regulation
Many studies involving in vitro, ex vivo (e.g., embryonic brain slices, which can also be subjected to focal electroporation[79]), and in vivo perturbations using single- or two-allele Zeb2 genetic inactivation (often cKOs) have been carried out (in mouse, about 50 in total). These KOs were initially based on the expression domain of Zeb2 in mouse embryos and early post-natal stages (see review Ref.[8]). These domains include the forebrain cortex, where Tcf4 is also expressed in all cells (be it at different intensities depending on the cortex layers, see below), and with Zeb2 transcripts and Zeb2 protein present in post-mitotic neurons of the upper layers, and both TFs are present in hippocampus. The KO-based studies in cultured cells and mice have also been combined with Zeb2 cDNA-based (again single- or two-allele) transgene cell-type specific rescue (involving cell-type specific Cre-controlled, from the Rosa26 locus with inserted Zeb2 cDNA). This allowed for the creation of various levels of Zeb2 in the model and obtaining detectable ranges of severity in cellular phenotype. Such experiments have contributed significantly to the conclusion that Zeb2 dosage, at the transcriptional level, needs precise control. Such control is necessary at every state/stage of Zeb2+ cells, for example, during exit from primed pluripotency (in ESCs[80]), but also in NPCs and other differentiating cell lineages in vivo, and/or terminal maturation of these (e.g., natural killer cells[81] and hematopoietic cell differentiation[82]).
ZEB2 expression is controlled through promoter-proximal (where TFs such as Smad2, ETS1, HIF1α, POU/Oct and NF-κB, E2F1, FoxQ1 and FoxA2, and Fra1/AP1 have been shown to bind) as well as distant regulations located upstream in the ~3.5 Mb-long gene desert (see Ref.[52], and references therein). In fact, ZEB2 ranks at the very top of proposed super-enhancer containing genes[83]. Interestingly, subtle differences in ZEB2 expression, in spatial-temporal terms and strength, and across species (e.g., human, gorilla, and chimpanzee, with its levels experimentally perturbed in the respective cerebral organoids), influence neuroepithelial transition (hence, also cell shape) and have been proposed to co-determine brain expansion in evolution[84]. Positive autoregulation of ZEB2 via a ZEB-binding site in the promoter-proximal region is an important driver of its upregulation during neural differentiation of pluripotent cells in vitro[64]. In addition, an adult-specific remote enhancer 165 kb upstream of the Zeb2 TSS that binds the bHLH TF E2A/Tcf3 via four E-boxes has been identified and experimentally proven to be critical with regard to the structural integrity of the Zeb2 locus and its regulation in adult (vs. fetal) hematopoiesis. In adults, this enhancer is essential for the production of monocytes, plasmacytoid dendritic cells (pDCs), and splenic B-cells[53].
Irrespective of the different TCF4 mRNA variants and protein isoforms and the resulting complexity, a detailed mapping of TCF4 presence in the brain remains critically needed to start correlating TCF4 dysfunction with pathophysiology in PTHS. Kim and Berens[85] constructed and validated a green fluorescent protein (GFP) reporter mouse model that produces a tagged TCF4-GFP protein under control of the otherwise unperturbed TCF4 locus. They specifically achieved this through the insertion of a splice acceptor-loxP-P2A-GFP-stop-loxP cassette between introns 17 and 18 (upstream of the bHLH domain encoding exons). In doing so, they documented TCF4 presence in the pallial region and cerebellum of postnatal brain. In the forebrain cortex, TCF4 is present in both glutamatergic and GABAergic neurons, as well as in astrocytes and mature oligodendrocytes. These observations, highly similar to those with ZEB2, strongly suggest that these latter cell types, and not only neurons, should also be studied when assessing TCF4 pathophysiology. Intronic SNPs in the TCF4 locus have been identified by GWAS, and one combination (rs9960767 with rs9960767) was found to correlate strongly with schizophrenia (SCZ) and-remarkably-improvement of verbal memory[86,87]. Jung et al.[88] systematically documented Tcf4 expression in the developing and adult mouse brain and provided strong support for the role of Tcf4 in the development and plasticity of cortical and hippocampal neurons. In doing so, they confirmed the structurally similar brain defects between Tcf4 mutant mice and PTHS patients. They also expanded their TCF4 localization studies to fetal and/or adult rhesus monkey and human brains and reported highly similar localization between these and mouse brains.
iPSC-derived PTHS patient NPCs and neurons and PTHS patient fibroblasts can also be used as models for studying PTHS[89]. Such cells also allow investigating how hypomorphic or dominant-negative mutations could cause PTHS. Genetic variation within the TCF4 locus or intronic SNPs may also cause ID in PTHS or associate with SCZ, respectively. Additionally, more detailed insight in TCF4 locus control may feed into pharmacological approaches to alter TCF4 expression in the CNS (and other tissues) and in the cellular models experimentally and document the effects via RNA-sequencing (RNA-seq).
EXPERIMENTAL PERTURBATION AND TRANSCRIPTOMIC PROFILING
Zeb2
Many reviews on ZEB2’s dynamic mRNA expression and ZEB2’s structure-function and role(s), including in the development of the nervous systems, have been published[8,90-93]. In short, in mice, Zeb2 cKOs (Δex7, targeted by Cre-recombinase and the critical and largest ex7[94]) have clearly confirmed Zeb2’s role(s) in early CNS development in mammals[79,95]. Most studies had their focus on the forebrain, but some also included mid- or hindbrain, or spinal cord, and neural crest-derived peripheral/enteric nervous systems (PNS/ENS; see review Ref.[8], and references therein). In the forebrain, studies expanded to early post-natal stages[79,96], adult neurogenesis[97], and injured brain[98], as well as in the PNS to (re)myelination[51,99] and pain sensing (see below).
In more recent studies, adult challenged mice have started to provide important roles of Zeb2 both as a marker and critical protective or causal gene (e.g., ulcerative colitis[100], heart infarct[101], liver steatosis and fibrosis[102], and melanoma[103]), thus, often in mouse systems/organs where no embryonic role or homeostatic role has been found, Zeb2 is not detectable or only at very low level expressed there or then. The recent increasing numbers of phenotypic descriptions of these challenged Zeb2-cKO mice, and in their organs or systems, may also urge clinical geneticists to follow up ontheir MOWS patients in the future in one or more of these extra directions.
Forebrain development
Good examples of Zeb2 functions in brain development, with clear consequences for MOWS, came from combining studies of forebrain cortex development and seizures/epilepsy, using a large panel of cortex[94] or interneuron specific Zeb2 KOs in the mouse forebrain[79,96].
Zeb2 deficiency results in insufficient numbers and misguidance of the ventral forebrain GABAergic interneurons caused by increased Dcc receptor and too high Unc5 receptor levels in these cells[79]. This has been proposed to interfere with long-range Netrin ligand-DCC receptor attractive cues for Unc5, and its levels do provide repulsive signals to Netrin. In the cortex itself, Zeb2 removal causes aberrant timing of cortical layer formation due to excess and precocious fibroblast growth factor (Fgf9) and neurotrophin (Nt3) production in the Zeb2+ upper cortex layers[95], which are secreted factors that feed back to the Zeb2-negative progenitor cells in the developing cortex. This observation, for the first time, identified a cell non-autonomous action mode of Zeb2, also in the entire field of brain corticogenesis, where TF-controlled neurogenesis in vitro in simple culture media recapitulated the successive waves of neurogenesis observed in vivo, leading to the assumption by most workers in the field that the action of the respective TFs was exclusively cell-autonomous.
Furthermore, bulk RNA-seq of Zeb2-cKO cells enriched from these respective models has also shown that intact Zeb2 is needed to precisely control genes involved in neuronal projection and connectivity, synaptogenesis, and synaptic plasticity[79], but these Zeb2 functions in these models remain understudied. Reduced levels of excitatory N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainic acid receptors in neurons, however, causes impaired Ca2+ signaling upon Zeb2 deficiency, again indicating a role for Zeb2 in neurotransmission and synaptic plasticity, possibly underlying late-onset epilepsy[104]. Further details on these specific phenotypes in these mouse models are again not (yet) available.
Progress may come here from using fused organoids of these cortical neuron types from the respective mouse mutants and eventually (im) balance between neuronal excitation and inhibition as a cause of epilepsy.
Neural crest-derived sensory neurons
Another interesting example, indicative of the role of Zeb2 in neural crest-derived cell types, is the reduced thermal pain response observed in adult Zeb2+/Δex7 mice, whereas mechanical pain was unaffected. This defect is due to reduced spike gain in capsaicin/heat-sensitive dorsal root ganglion (DRG) neurons only, accompanied by an upregulation of persistent Na+ channels and a decrease of delayed rectifier K+ channels. The signal propagation from the periphery to the spinal cord was affected as well in these mice. Zeb2 was therefore proposed to regulate thermal pain sensitivity by controlling the transduction properties of nociceptive primary sensory neurons in a novel manner, via changes in DRG voltage-gated ion channels[105].
The role of Zeb2 in inflammatory and neuropathic pain has been additionally investigated in adult Zeb2+/Δex7 mice challenged in various ways. Their hypoalgesic phenotype originates from the inflammatory component (hence, not from neuropathic pain) and depends on the Zeb2-controlled development of specific subsets of primary sensory DRG neurons (named C- and Aδ fibers)[105,106]. Therefore, the underreaction to pain observed in some MOWS patients is thought to result from a reduced responsivity to nociceptive stimulation rather than an inability to communicate discomfort. However, this remains, in general, difficult to investigate in these patients.
Other selected examples
A third selected example is the claimed association of ZEB2 with SCZ, as different SNPs located in close proximity to ZEB2 were found in different GWAS studies. One of these SNPs has been proposed to associate with two other psychiatric disorders, i.e., bipolar disorder and major depressive disorder[107]. These associations might explain some of the emotional disturbances observed in about one-third of MOWS patients and may, according to clinicians, reveal a risk of SCZ in these.
It is important to point out that Zeb2+/Δex7 mice do not develop histomorphological defects, as well as not at E8.5-9.5 stage, where embryonic lethality in Zeb2Δex7Δ/ex7 KOs is seen Ref.[94]. However, this may depend on genetic background[108] (see also Ref.[8]). Zeb2+/Δex7 adult mice in the appropriate background have craniofacial abnormalities, defects in specific brain regions (corpus callosum and a subclass of neurons in brain cortex), and interestingly (see below, grossly similar to Tcf4+/- mice) deviating behavioral activities, i.e., reduced motor activity, increased anxiety, and impaired sociability. Some of these defects are MOWS-like and PTHS-like and are not seen for Zeb2+/- mice, other closely related (ICR background) mice, and in outbred genetic backgrounds.
Novel avenues for studying the consequences of perturbed ZEB2 are now possible by deriving iPSCs from reprogrammed somatic cells of MOWS patients with precisely mapped mutation and submitting these cells in culture to neural differentiation over various periods of time (e.g., obtaining NPCs within few days and subsequently the subtypes of neuron as well). Considering many large ZEB2 deletions that are found in MOWS, the establishment of isogenic control iPSCs by correction of the mutation in patient iPSCs, as often strictly requested by the field, is a major problem. An alternative may be to reproduce such patients’ mutations by near-chromosome (as the length of the deletions) engineering in control iPSC lines. Milder cases of MOWS caused by subtle mutations may, because of the aforementioned need for isogenic, endogenous correction, then be investigated first in iPSCs. iPSCs can also be used for establishing longer-term cerebral (forebrain) organoids. Even if (some of) the ZEB2 cell non-autonomous functions would not be perfectly reconstituted in these systems, such iPSCs could still allow transcriptome analysis within ZEB2 (heterozygous) mutant pluripotent cells, their NPCs, and then derived neurons. The goal here is thus to identify MOWS signatures-or subsignatures, perhaps depending on the type of mutation-that ideally are relevant to neural development and/or could serve patient stratification.
Furthermore, comparing the transcriptome of such MOWS cellular systems with RNA-seq data of differentially expressed genes (DEGs) from the Zeb2-cKO mouse models is, in this respect, despite their striking intrinsic differences, already very meaningful. Moreover, such lists of DEGs in mutant cells can also serve as documentation of (which of these genes are) direct targets of ZEB2, and which of the latter encode for ZEB2 partner proteins, by applying ChIP-sequencing (ChIP-seq, or related strategies) for ZEB2 in combination with ZEB2 interactome studies.
Tcf4
Excellent overviews of the molecular mechanisms of TCF4 in PTHS, sometimes also addressing how these patients are treated and how TCF4 links to other psychiatric diseases, have been published (see[109], and references therein). Here, we focus on Tcf4 mutant mice and the consequences for embryonic and adult neurogenesis.
Adult neurogenesis
For adult neurogenesis, a recent observation was made with young adult mice of one of these Tcf4+/-models[110]. Indeed, in these mice, adult hippocampal neurogenesis is severely reduced, presenting as lower stem cell numbers and as impaired proliferation, maturation, and survival of adult-born neurons. Remarkably, these conditions severely ameliorate when the mice are housed long term in an enriched environment, previously known as a powerful stimulant of adult hippocampal neurogenesis (see review Ref.[111]).
Behavior and beyond
Tcf4+/- (more precisely Tcf4+/ΔbHLH) mice have been submitted to behavioral testing[112]. The ΔbHLH (Δ, deletion) approach was chosen because SNPs have been identified that generate a premature stop codon in or a missense mutant of this critical functional domain (i.e., different from other SNPs that associate with SCZ[113]). Based on observations with PTHS patients, the tests with these Tcf4+/ΔbHLHmice included motor/balance control and learning, spatial and associative learning and memory, assessment of autism, and extra tests. The Morris water maze, which can be combined with an object location memory task, is a classic hippocampus-dependent task that is used to evaluate spatial learning and memory. In doing so, significant deficits in spatial learning and memory were recorded for the Tcf4+/ΔbHLHmice. In the same context, hippocampus-dependent fear conditioning tests also revealed a strongly reduced freezing response. To assess autistic traits, the social interaction and repetitive behaviors were documented. These heterozygous mice favor social isolation over interaction, display increased grooming, are hyper-reflexive, and have deficits in prepulse inhibition, which all connect to SCZ and autism due to aberrant sensorimotor gating. The remaining tests revealed preferential use of more left vs. right front paw (when tested on the catwalk) and to put more weight (when tested for dynamic weight bearing) on left front paw. These mice also have weaker grip strength, remarkably in their hindlimbs only, and progressively higher activity in an open field and elevated plus-maze tests.
These already elaborate tests also encompassed RNA-seq on hippocampal CA1 tissue, identifying-based on comparisons that include Tcf4+/ΔbHLH cells-DEGs related to neuronal plasticity, axon guidance, cell adhesion (not via dysregulated Neurexins), Ca2+ signaling, and many upregulated neuroreceptors. The latter indicates that TCF4 (co-)determines the negative regulation of neurotransmitters in the hippocampus. Pathways in learning and memory are also downregulated (e.g., NMDA receptor), as well as those steering myelinations, while reduced global CpG-methylation was found across all gene bodies, but not in promoters. Changes in global demethylation, but here at both TSS and gene bodies, in these mutant mice are significantly linked with specific sets of upregulated genes. Taken together, Tcf4 haploinsufficiency in these mice clearly affects both expression and DNA-methylation of memory-relevant genes.
Corticogenesis
It is somewhat surprising that a detailed description of the brain-wide developmental defects in Tcf4 mutant mice has been lacking for quite some time. The defects include loss of pontine nucleus[114], promotion of differentiation of adult forebrain NSCs to neurons[115], and defects in neuronal migration and synapse formation in brain corticogenesis[116]. In addition, the careful mapping of timing and sites of Tcf4 mRNA expression, and ideally also the specific Tcf4 isoforms produced and the estimation of their quantity, is still needed as part of the detailed phenotypes, beyond the admirable efforts already dedicated to this profiling of complex transcripts in the mouse[71]. In any case, Mesman et al.[117] demonstrated the presence of Tcf4 mRNAs throughout the mouse brain from E12.5 onwards, but at different intensities: Tcf4 mRNA levels were found highest in E14.5 cortical plate (CP) of the forming forebrain cortex, and then also at E16.5 in CP and intermediate zone, demonstrating layer-specific Tcf4 mRNA levels. At the protein level, this layer-specific presence could be confirmed from E16.5 onwards, a time point where most neurons of the developing cortex have been specified. Tcf4 locates to cells (but not all cells) of layers V and VI, and only very low staining of Tcf4 is detected in the ventricular and subventricular zone (VZ and SVZ), possibly within cells that have already acquired a neuronal fate.
This same study also impressively combined Tcf4+/- and Tcf4-/- mice in the analysis of defects in brain cortex architecture. CP thickness is reduced, while at P0, the layers in both mutant mouse models are present, and cell numbers positive for markers for layers V and VI are there (and differ between the two mutants). The cortex layers are also less distinctive, possibly due to aberrant neuronal differentiation and defective segregation of Ctip2+ (corticospinal) and Satb2+ (callosal projection) neurons between E17.5 and P0. There is an indication of the delayed formation of Ctip2+ cells in particular, but also at P0, as based on double-positive Ctip2+/Satb2+ cell numbers. This suggests severe misspecification of the cortical layers, and this has a dramatic impact on the presence of Cux1, which in the mutants is now absent in the upper layers of the Tcf4-/- mice but is intact in the VZ/SVZ in all mouse genotypes studied here.
Additional thorough phenotyping also showed that loss of Tcf4 causes defects in the development of the corpus callosum, the anterior commissure, and hippocampus (at E17.5), which persist until birth. Early phenotyping by RNA-seq of the early E14.5 cortex identified ~130 downregulated genes. These confirm Tcf4 as a candidate for direct transcriptional activation of genes steering neuronal differentiation and migration and included NeuroD1, Mash1, and Id2. Interestingly, the downregulated genes also encompassed genes related to SCZ or synaptic plasticity. Taken together, many of the defects observed in PTHS patients were recapitulated in the mutant animals, with fairly mild impact in the Tcf4+/-, but more dramatic effects in the Tcf4-/- mice.
A complicating factor here is possibly that different teams have targeted different critical Tcf4 exons and/or replaced these with a (non-removed) neomycin resistance cassette. Therefore, other mouse models have proven extremely helpful and informative. Thaxton et al.[118] designed two precise Tcf4 mutant mice, i.e., a human R580W model (a most common mutation in PTHS, corresponding to mouse R579W) and a model that deletes in-frame three pathogenic R-residues. These mice were compared to ubiquitous as well as CNS-specific Tcf4+/- mice. They display common, consistent pathophysiology with regard to microcephaly, deficient spatial learning and anxiety, hyperactivity, and, when comparing some of these mice further, NMDA receptor hyperactivity in the hippocampus. A CNS-cKO (using hGFAP-Cre;Tcf4fl/fl) was established[119] and allowed the study in adult mice of the consequences of complete Tcf4 removal. This CNS cKO Tcf4 mouse confirmed the previously documented phenotypes, but also delivered new information, in particular on abnormal neuronal morphology (shortened apical dendrites, increased branching of dendrites).
Myelinating cells, also in syndromic autism disorders
For both Tcf4 and Zeb2[84,97], neuronal shape and morphology have clearly emerged as a new topic of interest. As for Zeb2, studies of the role of Tcf4 and how PTHS affects non-neuronal cells in the CNS have been initiated and include oligodendrocyte differentiation and myelination. One study, using five different appropriate PTHS models that show cell-autonomous reductions in oligodendrocyte numbers and myelination, and two extra syndromic autism disorder mouse models (Pten and Mecp2), showed in an impressive way myelin-related transcriptomic profiles that are shared between PTHS and human autism disorders[120]. An elegant step to get to this result was to combine the PTHS mouse model DEGs with RNA-seq data from post-mortem autism human brains. As a whole, such studies increasingly confront autism researchers with decisions to embark on CNS myelinogenesis and myelination research. Indeed, the RNA-seq results obtained in the aforementioned multiple Tcf4 mutants[120] reveal in the adult brain age-specific DEGs affecting processes in forebrain development, neuron projection, and axon development; post-synaptic density for the upregulated genes in the mutants; and axon ensheathment and myelination for the downregulated genes[120]. The RNA-seq data thus prompted the validation of myelination deficits in the PTHS mouse models ex vivo and in vitro, by marker analysis of both oligodendrocyte precursor cells (OPCs) and their differentiated progeny. These experiments were expanded with microscopy to document myelination and with electrophysiology to record the propagation of compound action potentials. They indicated that PTHS mice have reduced myelinated axons, as also seen in specific homozygous Zeb2-cKO mice[51,99,121], resulting in a greater proportion of neuronal activity being transmitted down clearly under- or even unmyelinated axons.
Extra experiments in one of their specific PTHS mouse models (i.e., Tcf4+/tr, where trTcf4 is proposed to be dominant-negative) clearly indicated that, although the in vitro produced OPCs were higher in number than in wild-type Tcf4+/+ control mice, they still produced fewer oligodendrocytes. Thus, Tcf4 regulates OPCs, joining Zeb2 as a critical factor, for promoting OPC differentiation into myelinating cells in the embryonic CNS (and the postnatal PNS). Meanwhile, Tcf4 function in OPCs has been shown via additional Tcf4 cKO mice to be critical for medial ganglionic eminence (MGE) and anterior entopeduncular area (AEP)-derived OPCs, the first source of OPCs destined to the olfactory bulb (OB)[122]. In line with the aforementioned observations, conditional genetic inactivation (using Nkx2.1-Cre) of Tcf4 causes increased numbers of OPCs in the OBs. Furthermore, Tcf4 suppresses apoptosis by mediating survival in a cell-autonomous fashion (as shown by cell transplantation assays), with apoptosis being controlled by Bax/Bak, Bcl2 family members that are within the nucleus core regulators of the intrinsic pathway of apoptosis, the classical form of programmed cell death, accompanied by mitochondrial herniation. In these mice, it was important to show that the differentiation of (e.g., Lhx1+ and Dlx1+) NPCs remains unaffected in the MGE and AEP upon the loss of Tcf4.
Immune cells
As in the case of Zeb2, the role of Tcf4 is, of course, not limited to neurodevelopment and brain function. Tcf4 is largely dispensable for T cell and B cell development in the mouse. However, PTHS patients sometimes display impaired interferon (IFN) response and deviating expression profiles in pDCs, a distinct subclass of dendritic cells that rapidly produce type I IFNs in response to virus infection. Elegant transplantation studies using Tcf4 KO mouse fetal liver cells into lethally irradiated recipient mice, followed by the analysis of the appropriate donor-derived hematopoietic cells, showed efficient reconstitution, giving rise to myeloid, T and B, and DC subsets. However, Tcf4-KO derived pDCs were missing from the bone marrow and all lymphoid organs, and splenocytes and the bone marrow cells failed to produce IFNα.
These important results were followed by further studies of the consequences of tamoxifen (TAM)-inducible Tcf4 KO in mice, with a focus on the analysis of bone marrow and spleens. These studies confirmed the results and also showed that only Tcf4 has a specific role (unlike two other tested E-family proteins), acts cell-autonomously in pDC development, and displays haploinsufficiency in mice. Thus, Tcf4 removal in HSCs/HPCs results in failure of pDC development, identifying Tcf4 as an essential TF in the innate immune system in mice as well as humans. Indeed, in PTHS patients, marker protein deviations and functional defects of pDCs have been documented, boosted by these results in KO mice[123].
Cell culture studies
As for ZEB2, other avenues using cultured cells are being used to improve our understanding of TCF4 action in healthy and PTHS brains. Experimental KD of TCF4 by siRNA has been used in both cell lines and NPCs, but initially not always of (fetal) forebrain origin. One study used a conditionally (TAM-induced) proliferative and immortalized (via a c-MycERTAM transgene) cell line from human fetal cortical neuroepithelium, cells of which can differentiate into neurons and astrocytes. They performed successful TCF4 siRNA experiments (using two siRNAs) in these cells that were carefully carried out in the absence of TAM[124]. Microarray-based transcriptome analysis identified about 1750 and 1500 microarray probes, respectively, that differed as compared to control siRNA, with ~630 of these behaving similarly in the two TCF4 siRNA setups. Candidate genes (39 in total) that survived a stringent analysis, and thus presented as a high-confidence set, were highly enriched for genes involved in cell cycle control.
This same work[124] also involved the testing for association with SCZ (using large-scale GWAS results from others), leading to five out of nine of those genes to the most significant association (i.e., GNL3, STAG1, CENPM, NCAPD3, and CDC20), again pointing at cell cycle control. However, while the emphasis of this study is on cell cycle genes, the identified shared DEGs also included genes related to various other functions in the embryonic and adult brain. A second, similar perturbation study, using acute TCF4 KD in the neuroblastoma cell line SH-SY5Y followed by RNA-seq, identified ~1200 DEGs (~500 genes up, ~700 genes down)[125]. Over-represented genes here are genes involved in the TGFβ-family signaling pathway, epithelial-to-mesenchymal transition (EMT), and apoptosis. Relevant candidates to follow up on are SNAI2, NEUROG2, and ASCL1; the syndrome-causing genes UBE3A and MEF2C; and, not unexpected, ZEB2.
An intermediate approach between in vivo and cell culture experimentation, again involving Tcf4 perturbation, has also been used, i.e., in utero electroporation of the cerebral cortex area of E14.5 mice using Tcf4-shRNA combined with tracer GFP vectors[126]. Analysis at P0 identified accumulation of GFP+ cells in the IZ and VZ instead of migrating to the CP, as happens in the controls. Thus, normal Tcf4 levels are essential for neuronal migration, with likely an underlying aberrant neuronal morphology due to the absence of a leading process, which is already visible at E17.5. The authors excluded that the defect arose from aberrant radial glia organization, division, or survival. Importantly, the Tcf4 KD in the neurons causes upregulation of Bmp7, of which the proximal promoter has multiple E-boxes, with two of these shown to bind Tcf4. These observations make Bmp7 an important effector (that is negatively regulated by Tcf4) of neuronal migration. This was further substantiated by additional experiments involving electroporation of expressible Bmp7-encoding vectors into control mouse embryos. These results confirm the contribution of Bmp7 to the phenotype on the one hand and (partial) rescue of the Tcf4 KD cells obtained by electroporation of Bmp7-shRNA on the other hand.
TARGET GENES AND INTERACTOMES
In addition to a meticulous description of phenotypes caused by ZEB2 and TCF4 deficiency in cellular and mouse models and patients, decisive insight into the action mechanisms of these TFs will come from the mapping of their genome-wide binding sites and the identification of co-operating partners or complexes (using proteomics), preferably in neural cells.
Mapping of genome-wide binding sites
No satisfying anti-Zeb2 antibodies are available for efficient ChIP-seq. Recently, mouse ESCs were edited (using CRISPR-Cas) that produce tagged Zeb2-V5 (V5 heterologous epitope) from one of its two endogenous alleles[64]. ChIP-seq (using anti-V5 antibody) mapped ~2400 Zeb2 binding sites in ESC-derived NPCs, with ~2300 of these annotated to ~2000 protein-encoding genes. Zeb2 is mainly recruited to the promoter-proximal region of transcription regulator or protein modifying enzyme encoding genes, but it also binds to distant REs. The ChIP+ genes include genes classified in Wnt signaling, which is in line with previous observations of DEGs of the Wnt system in Zeb2-cKO (CNS) mouse models. A second group included Dnmt3 family genes, which is in line with previous observations in Zeb2-KO ESCs submitted to neural differentiation[80]. Of the aforementioned 2000 genes, about 1200 are differentially expressed during neural differentiation in vitro on Day 4 (epiblast-like stem cells) and Days 6 and 8 (NPCs) as compared to Day 0 (ESCs), including Tcf4. Importantly, Zeb2 was also found to potentiate its own expression during NPC production via a promoter-proximal binding site. This autoregulation, as shown by homozygous deletion of the ChIP+ DNA segment (again using CRISPR-Cas), is crucial for proper control of many of its key ChIP+ target genes in ESC-based neural differentiation[64].
ChIP-seq for tagged V5-Tcf4 in mouse NSCs showed Tcf4 binding to enhancers and super-enhancers[83], including loci encoding neurogenic bHLH factors, but also-albeit not yielding a strong signal in NSCs-its own promoter[78]. Tcf4 was found to bind to its targets together with its interaction partners such as Smad4, Sox2, and Chd7[14]. Gene ontology analysis on Tcf4 target genes, i.e., bound and activated by Tcf4, showed that Tcf4 predominantly controls genes involved in the cell cycle and M-phase of the latter. This could fit with the microcephaly phenotype that is part of PTHS.
Interactomes
Transitions of stem cells into different lineages, and progression of cell differentiation and maturation, including neural cell types, are driven by changes in gene expression regulated by specific TFs that often bind to evolutionary conserved DNA motifs in TSS-proximal sequences (e.g., promoters and first intron) and to distant REs. Transcriptome, epigenome, and higher-order chromatin do not stand by themselves but are intertwined and influence each other herein. This, in collaboration with the actions of chromatin-modulating complexes, results in the proper activation or repression of target genes. Chromatin modulation involves re-modelers and modifying enzymes including methyltransferases, HATs and kinases (often collectively called chromatin writers), demethylases and histone deacetylases (HDACs, erasers), and complexes such as the switch/sucrose nonfermenting (SWI/SNF) complex [also known as BRG1/BRM-associated factor (BAF complex); named readers]. Enhancers are often located in-cis kb to sometimes Mb away from the specific genes they regulate, requiring TF- and Mediator (complex)-assisted DNA-looping. Thus, correct gene expression also requires epigenetic reorganization via chromatin modifications and chromatin remodeling.
Already from the earliest studies with Zeb1, ZEBs have been proposed as putative competitors in DNA-binding for E-box-binding transcriptional activators (bHLH TFs) simply by target-site occupancy, soon followed by the first reports of ZEBs being active transcriptional repressors in most assays. For this latter activity, co-repressor binding has proven important, e.g., with NuRD. It is composed of, among others, at least the ATPases CHD3/4/5, HDAC1/2, metastasis-associated protein (MTA)-1/2/3, retinoblastoma binding protein and histone chaperone RbBP-4/7 (also named RbAp48/46), methyl-CpG-binding domain protein (MBD)-2/3, and zinc-finger TFs GATA2A/2B[127,128]. For repression, this complex thus combines deacetylation of histone-H3 by HDAC1/2 with demethylation of H3K4 (see review Ref.[129]). NuRD-mediated chromatin remodeling in cerebral cortex development, neurodevelopmental disorders, and myelination was recently reviewed[6,29,130,131].
Various subunits of the NuRD complex, including CHD4, were identified in affinity-purified Zeb2 complexes isolated from Zeb2-overproducing heterologous cells[50]. ZEB2 associates with NuRD via its NIM (see above), but the subunit/s of NuRD that mediates this direct interaction is/are not known. In two aforementioned patients with mild MOWS caused by mutations affecting the ZEB2 NIM, one being a subtle R22G missense mutant, the only direct molecular detectable consequence is that ZEB2 interaction with the NuRD complex is lost. Such aberrant ZEB2 is indeed unable to recruit NuRD (subunits) but, as a result, displays reduced transcriptional repression activity on, e.g., the XBMP4 gene promoter (in Xenopus (X) embryos), a target of XZeb2. By using a dominant-negative variant of the ATP-binding NuRD component CHD4/Mi-2 (its in vitro made sense RNA was injected in 1-4 cell stage Xenopus embryos), it was shown that this subunit and thus NuRD is actively involved in repression of the ZEB2 target gene Cdh1 (encoding E-cadherin, cadherin-1) and in XZeb2-controlled neural induction[50]. This work also showed that the absence of NuRD binding to human ZEB2 can be the cause of mild forms of MOWS. Similar functional consequences (and inability to rescue Zeb2-KO) of NIM-mutant Zeb2 were documented in myelination and adult neurogenesis[51,97]. Cellular systems that enable documenting GWBS for a NIM-defective human ZEB2 are not available yet; hence, target genes that may co-depend or depend exclusively on NuRD-ZEB2 interaction remain unknown.
Tcf4 interacts with more than 100 partners[14], including chromatin remodeling complexes such as SWI-SNF, NuRD, and TRRAP, but also many TFs. Known partners, including HLH factors Ascl1 (also named Mash1), Id1, and Id3/4, were also identified in this study. Other bHLH factors (Tcf12 and E2-A/Tcf3) bind to Tcf4 as well. Interestingly, Zeb2 was detected in tagged Tcf4, Sox2, and Npas3 interactomes[14]. A major functional finding here was that Tcf4 binds and activates multiple genes involved in primary microcephaly, such as Mcph1 and Wdr62. It co-localizes on primary microcephaly genes with its interaction partners Smad4, Sox2, Chd7, and Ep300. Smad4 also (co-)activates transcription of Mcph1 and Wdr62. Mutations in SMAD4, EP300, CHD7, and SOX2 cause microcephaly in at least some patients. Therefore, a picture has emerged that TCF4 is part of a protein interaction network regulating human primary microcephaly genes, in which network members cause microcephaly as part of the human phenotype they cause.
Mediator was not picked up in Tcf4 co-purifications[14], but vice versa Tcf4 did show up prominently in purification of the Mediator complex from NSCs[78]. Identification of enhancers and super-enhancers in NSCs by Mediator ChIP-seq showed that they nearly completely overlap with genome-wide binding of Tcf4. Moreover, Tcf4 KD lowered Mediator binding to common binding sites, showing that Tcf4 exerts genome-recruitment activity on Mediator[78]. This co-localization of TCF4 and Mediator is striking, and in fact, goes beyond enhancers, making both Tcf4 and Mediator mark so-called broad promoters[132,133]. Broad promoter containing genes are strongly linked to cell identity. This suggests that the identified Tcf4-Mediator axis plays a major role in NSCs[78].
Impact on or by Wnt signaling
The most affected deregulated genes resulting from Zeb2 deficiency, certainly in the development of the nervous systems, but also in cell culture, often map to the Wnt signaling system. This is the case in mouse hippocampal development[134], embryonic CNS myelinogenesis, and post-natal Schwann cell (SC)-mediated (re)myelination in the PNS[51,99,121]. Myelination (see also above) is absolutely required for optimal conduction velocity of nerve impulses, and defects may cause disrupted motor function. It occurs by the formation of myelin sheets generated by oligodendrocytes/SCs, which derive from OPCs. The differentiation of these OPCs toward mature, myelinating cells is inhibited by BMP, Wnt, and Notch signaling[135]. Zeb2 protein is present in cells that accompany motor neuron axons in the trunk of early mouse embryos at the level of the limbs[136]. Zeb2 was proposed as a strong direct target for the upstream TF Olig2, and Zeb2 mRNA requires normal levels of Olig1. Olig1 and Olig2 (including their overproduction) also impact neurodevelopmental disorders, even in Down Syndrome and autism spectrum disorders[137]. Furthermore, Olig1 and Olig2 bHLH TFs are essential for CNS myelinogenesis[137]. Analysis of Olig1 and Olig2 KO mice revealed that in P14 and E14.5 spinal cords, respectively, Zeb2 mRNA levels become extremely low to undetectable. In cultures of adult rodent early-OPCs, overproduction of Olig1 and/or Olig2 activates Zeb2. High levels of Zeb2 protein are present in mature oligodendrocytes, and only very low levels are detected in OPCs. These observations provided a strong basis for generating Olig1-Cre;Zeb2 cKOs and documenting the effects of Zeb2’s removal on CNS myelinogenesis. Such mice are viable, but two weeks after birth, they develop generalized tremors, limb paralysis, and seizures, similar to the symptoms observed in other genetic mouse models with defective myelinogenesis.
Intact Zeb2 was further shown to be dispensable in platelet-derived growth factor receptor-alpha (PDGFRα)+ OPCs, but its presence is critical for their differentiation. Subsequent experiments demonstrated that Zeb2 downstream of Olig1 and Olig2 is needed during CNS myelinogenesis for differentiation of OPCs to myelinating cells, and that this requires both Zeb2’s repressor and activator activities[8]. Importantly, Zeb2 represses sets of BMP-Smad-activated genes that normally inhibit myelinogenesis, and Zeb2 directly activates Smad7, which (co-)downregulates BMP signaling with Zeb2 (via BMP-pSmad interaction). In a Smurf-dependent manner, Smad7 also sends Wnt-activated β-catenin (βcat), with the latter normally inhibiting myelinogenesis also, into degradation[121]. Thus, in a cell-intrinsic manner, Zeb2 is needed to generate anti-BMP-Smad activity and anti-Wnt-βcat effects in CNS myelinogenesis.
Based on this, a number of predictions could be tested and confirmed. One is that, in neonatal OPCs, whose proliferation is stimulated by PDGF-AA, Zeb2 overproduction would enhance their differentiation to mature oligodendrocytes, as detected by downregulation of genes encoding negative regulators of differentiation, and a strong increase of Sox10 and Olig2 expression. Furthermore, other experiments demonstrated that Zeb2-P300 co-operation is responsible for direct activation of the pro-myelination genes. Another prediction was that Smad7 overproduction would rescue the myelination defect in Zeb2 KO OPCs, which was the case[121]. These results are the start of new investigations aiming at addressing the Zeb2 function in SCs of the adult PNS, where Zeb2 is present[51]. Sciatic nerve injury already rapidly upregulates Zeb2 (in a few hours), and Zeb2 levels remain high in remyelinating SCs for a long time after the injury[99].
Taken together, these Zeb2 studies of the cellular and molecular phenotypes in CNS myelinogenesis and PNS myelination are comparable in many respects. They show that intact Zeb2 is needed to control signaling pathways that impact myelinating cells, i.e., Zeb2 is needed to generate and maintain anti-Notch/Hey2 and anti-Sox2, as well as (in both CNS and PNS) anti-BMP and anti-Wnt activities in these cells. Rescue experiments with the NIM-deficient Zeb2R22G mutant (see above) highlighted the importance of NuRD-interaction. Importantly, these results in the mouse models recapitulate the phenotypes observed in MOWS patients presenting a delay in myelination and defects in white matter, proposed to cause motor deficiencies such as a delay in walking.
Modulation of Wnt/βcat active signaling and epigenetic status also caught attention by the TCF4 field[138]. In the case of signaling, the approaches used Wnt pathway activation via the addition of Wnt3a-containing conditioned media or CHIR-99021, a potent inhibitor targeting GSK3, wherein GSK3 normally negatively regulates canonical Wnt signaling. Both treatments resulted in 1.4-6-fold upregulation of TCF4 depending on the specific transcripts analyzed, but these are experimentally difficult to allocate via Western blotting, due to the different TCF4 protein isoforms (see above). Further analysis, also involving the annotation of potential TCF/LEF binding sites in TSS-proximal sequences of Wnt-responsive genes, revealed that the highest Wnt-responsive TCF4 transcripts [encoding a short form of TCF4, TCF4-A; see Figure 1C] contained multiple TCF binding sites, which could-albeit indirectly-be confirmed for binding (i.e., by ChIP-seq of TCF7L2).
A next approach by the same authors used two different pharmacological inhibitors of Wnt canonical signaling in NPCs stimulated with Wnt-containing conditioned medium (CM) (i.e., the tankyrase inhibitor XAV-939, causing enhanced degradation of βcat, and ICG-001, preventing βcat from binding to the HAT CBP, a member of the P300 family). These treatments were found to downregulate TCF4 mRNA expression. They also paved the way to test selective HDAC inhibitors, investigate, in particular, the exclusive synergism of Class I HDAC inhibitors in Wnt-CM conditions only, and document these experiments by RNA-seq and TCF4 binding site analyses, in particular for DEGs[138].
Many of these DEGs are known as critical to nervous system development and function and act in synaptogenesis, post-synaptic density scaffolding and signaling, and axon growth and guidance. In addition, many of these DEGs and other ones have been associated with autism, ID, and/or psychiatric disorders. They represent interesting downstream markers and are part of signatures that can be compared between PTHS patient-derived cells and neurons. Such work is also inspiring for ZEB2 research into pharmacological modulation that targets ZEB2 expression regulation from the single remaining intact ZEB2 allele in MOWS.
ZEB2 and TCF4, each other’s modifier for neural differentiation in vitro, but also of other genes in normal brain development?
As illustrated above for ZEB2 and TCF4, but also applicable to other neurodevelopmental TFs, the combination of genetic analysis and mutation mapping, the phenotypic and transcriptomic analyses of appropriate cellular and animal models, and the mapping of genomic TF binding-sites and TF interactomes give insight into TF actions (also in concert with chromatin remodeling complexes) at the level of individual disease genes, as well as GRNs and PRNs, including these steering neural differentiation.
The first example of new insight is that a number of Zeb2 ChIP+ target genes, at least as mapped thus far in mouse ESC-derived NPCs, could be associated with human neurodevelopmental disorders (such as ID, mental disorders, eye defects, seizures, and speech impairment). For instance, a strong Zeb2 binding site has been mapped to the PTHS Tcf4 gene proximal region[64]. In addition, in the same set of experiments, perturbation of Zeb2 autoregulation via mutating the Zeb2-binding site in the promoter-proximal region of Zeb2 results in strong downregulation of not only Zeb2 but also Tcf4. This DNA segment, however, remains to be tested firmly for Tcf4 occupancy. Preliminary observations suggest this signal is not strong, at least in NSCs, possibly as a result of the combination of Zeb2 being expressed at a low level and Tcf4 having a preference for enhancer binding, including its own enhancer located 200 kb within the gene[78]. Thus, by extension, in NSCs and early neurogenesis, the aforementioned possibility of competition between Zeb2 and a Tcf4-bHLH heterodimer for E-boxes is unlikely, although this still has to be investigated in depth for competition at the Zeb2 autoregulatory site.
Obviously, one still needs details on enhancers as well as meaningful SNPs for both TCF4 and ZEB2, and how variation in those affects the levels of their mRNA and protein levels including in, e.g., human NPCs, as well as how this affects for each factor its interactions. In more general terms, this also means that, in neurodevelopmental iPSC-derived models, the effect of a perturbation of a given TF or chromatin remodeler will have to be documented in terms of its epistasis or GRN/PRN. This can occur via KD or, alternatively, via mutation in a domain or allelic genetic inactivation. The choice of KD vs. KO may influence epistatic relations and compensatory pathways, which may differ between choices. In any case, such sophisticated biochemical and functional studies are expected to confirm ZEB2 and/or TCF4 as important hubs in GRNs/PRNs, but also as modifier gene(s). They could very well be modifiers not only of one another (empowered by similarities of defects between MOWS and PTHS), but also of other causal genes in phenotypically overlapping neurodevelopmental syndromes (discussed in Ref.[64]).
DIAGNOSIS AND STANDARD THERAPIES
A diagnosis of MOWS or PTHS is usually not made immediately after birth, but rather during infancy or childhood. It is based on thorough clinical evaluation, identification of characteristic physical features such as facial appearance (certainly for MOWS), and other symptoms, which do not generally present with all patients and, in the case of MOWS, clearly range from severe to milder cases. Of course, conditions of HSCR (for MOWS, but constipation is also very common in PTHS) and seizures (for both MOWS and PTHS) are also important indicators[8.9,41,46]. In addition, for both syndromes, the structural and functional features become more pronounced with time; thus, diagnosis is easier in older patients. Checking for defects also includes imaging techniques[45] such as computerized tomography scanning or magnetic resonance imaging of the brain and ultrasound of other organs (heart and kidney). Obviously, the clinical diagnosis is best confirmed by molecular genetic testing for mutations in the ZEB2 and TCF4 genes, respectively, with the current focus thus far on exon-sequencing, and it can be accompanied by the firm exclusion of other related disorders for which the causal genes (e.g., for ID) are known.
Currently, we can cure neither MOWS nor PTHS. The remarkably multiple functions of these TFs during embryogenesis and in the entire postnatal body, the knowledge of the precise regulation of ZEB2 and TCF4 levels in various cell types as concluded from studies of their gene expression control or via miRNAs (including lncRNAs, some of which act as sponges for, e.g., ZEB2-targeting miRNAs), and their complex interactome do not yet provide a basis solid and/or understood enough for drug-based or targeted protein-protein interaction-based intervention. Conversely, any attempt to increase ZEB2 levels, which might be considered in MOWS (experimentally by gene- or RNA-based therapy), holds many dangers. For example, the resulting overproduction of ZEB2 impacts on, e.g., pathological EMT (see the recent discussion[139]) and correlates with bad prognosis in several epithelial-derived cancers, including melanoma[103] and gastric cancer[140].
The current treatment of MOWS and PTHS patients is directed toward the specific symptoms, and hence needs, of each affected individual and requires a team of specialists. These teams range from neurologists (prescribing anti-epileptic medication, which for example, in MOWS appears more easily managed in adolescents and adults) to gastroenterologists, dieticians, ophthalmologists, sometimes surgeons (in the case of HSCR in MOWS), pulmonologists (in PTHS, where the onset of breathing abnormalities can vary from months to years after birth), cardiologists, and psychologists to work together and case-by-case tailor the treatment plan. Treatment of bowel obstruction and HSCR involves complex surgery, often over several interventions, including colostomy and thereafter the removal of the non-functioning section of the colon combined with rejoining the healthy segments of the bowel. Other surgeries may also be performed to treat specific congenital anomalies, such as heart defects.
Physical, occupational, and speech therapies are very useful in helping children with developmental delay to reach a higher potential, while coordinated establishment of patient registries, genetic mapping, and longitudinal follow-up of patients are very valuable. As argued here and elsewhere[8], research in MOWS- and PTHS-like animal models (in particular, in challenged adult mice) in many biomedical fields is likely to inspire clinicians to follow up on their, e.g., injured (reduced pain sensitization, in MOWS) and aging patients more broadly, certainly in the case of complicating acute (including infections and immunity) or chronic (liver fibrosis[102] and myelinating diseases[51,99]) pathology.
CONCLUSION
It is amazing how investigation of mechanisms underlying monogenic syndromes, in particular by establishment of animal and cellular models combined with multi-omics approaches, has rapidly complemented ongoing parallel efforts in studies in clinical genetics, establishment of patient registries, and execution of genetic testing. At the same time, this combination of fundamental and clinical research provides insight into phenotypic overlap and possibly convergence between different syndromes. Here, we suggest this is the case for MOWS (ZEB2) and PTHS (TCF4), prompting us to summarize recent progress on both and combine them for the first time in this review. The progress is expected to provide a solid basis for designing next-generation cellular and animal models and use these to map transcriptomic signatures for each of the applied (including domain and patient-inspired) mutations of these TFs. We also highlight the importance of re-addressing their gene expression regulation (including via distal regulatory elements) and documenting and following up on patients with regard to non-neuronal cells of their nervous systems, as well as outside of these systems when these patients suffer certain acute or chronic pathologies.
DECLARATIONS
Acknowledgments
We thank all workers of the Center for Biomics-Genomics for sequencing and bio-informatics, Mike Dekker and Anne Korporaal for expert technical assistance, and Niels Galjart and Frank Grosveld for discussions. We are grateful to the clinical input from Livia Garavelli and Christiane Zweier.
Authors’ contributions
Carried out most of the TCF4 and ZEB2 studies in the NPC models: Meert L, Birkhoff JC, Conidi A
Co-designed and supervised these projects: Poot RA, Huylebroeck D
All authors contributed to and approved the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by Erasmus MC via departmental and BIG project funding (together with Erasmus University Rotterdam).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
REFERENCES
1. Hempel A, Pagnamenta AT, Blyth M, et al. DDD Collaboration. deletions and de novo mutations of sox11 are associated with a neurodevelopmental disorder with features of coffin-siris syndrome. J Med Genet 2016;53:152-62.
2. Al-Naama N, Mackeh R, Kino T. C2H2-type zinc finger proteins in brain development, neurodevelopmental, and other neuropsychiatric disorders: systematic literature-based analysis. Front Neurol 2020;11:32.
3. Zhang K, Yu F, Zhu J, et al. Imbalance of excitatory/inhibitory neuron differentiation in neurodevelopmental disorders with an NR2F1 point mutation. Cell Rep 2020;31:107521.
4. Blok L, Rousseau J, Twist J, et al; DDD study. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat Commun 2018;9:4619.
5. Yasin H, Gibson WT, Langlois S, et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J Hum Genet 2019;64:271-80.
7. Mossink B, Negwer M, Schubert D, Nadif Kasri N. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci 2021;78:2517-63.
8. Birkhoff JC, Huylebroeck D, Conidi A. ZEB2, the Mowat-Wilson syndrome transcription factor: confirmations, novel functions, and continuing surprises. Genes (Basel) 2021;12:1037.
9. Teixeira JR, Szeto RA, Carvalho VMA, Muotri AR, Papes F. Transcription factor 4 and its association with psychiatric disorders. Transl Psychiatry 2021;11:19.
10. Chen ES, Gigek CO, Rosenfeld JA, et al. Molecular convergence of neurodevelopmental disorders. Am J Hum Genet 2014;95:490-508.
11. Sugathan A, Biagioli M, Golzio C, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A 2014;111:E4468-77.
12. Engelen E, Akinci U, Bryne JC, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 2011;43:607-11.
13. Maussion G, Diallo AB, Gigek CO, et al. Investigation of genes important in neurodevelopment disorders in adult human brain. Hum Genet 2015;134:1037-53.
14. Moen MJ, Adams HH, Brandsma JH, et al. An interaction network of mental disorder proteins in neural stem cells. Transl Psychiatry 2017;7:e1082.
15. Estruch SB, Graham SA, Quevedo M, et al. Proteomic analysis of FOXP proteins reveals interactions between cortical transcription factors associated with neurodevelopmental disorders. Hum Mol Genet 2018; doi: 10.1093/hmg/ddy035.
16. Gabriele M, Lopez Tobon A, D’Agostino G, Testa G. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog Neuropsychopharmacol Biol Psychiatry 2018;84:306-27.
17. Lewis EM, Kroll KL. Development and disease in a dish: the epigenetics of neurodevelopmental disorders. Epigenomics 2018;10:219-31.
18. Fallah MS, Szarics D, Robson CM, Eubanks JH. Impaired regulation of histone methylation and acetylation underlies specific neurodevelopmental disorders. Front Genet 2020;11:613098.
19. Cogné B, Ehresmann S, Beauregard-Lacroix E, et al. CAUSES Study; Deciphering Developmental Disorders study. Missense variants in the histone acetyltransferase complex component gene TRRAP cause autism and syndromic intellectual disability. Am J Hum Genet 2019;104:530-41.
20. Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol 1995;168:342-57.
21. Okabe S, Forsberg-nilsson K, Spiro AC, Segal M, Mckay RD. Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro. Mechanisms of Development 1996;59:89-102.
22. Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 2001;19:1129-33.
23. Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 2003;21:183-6.
24. Peljto M, Wichterle H. Programming embryonic stem cells to neuronal subtypes. Curr Opin Neurobiol 2011;21:43-51.
25. Eiraku M, Sasai Y. Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr Opin Neurobiol 2012;22:768-77.
26. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 2014;9:2329-40.
27. Zhang M, Ngo J, Pirozzi F, Sun YP, Wynshaw-Boris A. Highly efficient methods to obtain homogeneous dorsal neural progenitor cells from human and mouse embryonic stem cells and induced pluripotent stem cells. Stem Cell Res Ther 2018;9:67.
28. Galiakberova AA, Dashinimaev EB. Neural stem cells and methods for their generation from induced pluripotent stem cells. in vitro ;8:815.
30. Doostparast Torshizi A, Armoskus C, Zhang H, et al. Deconvolution of transcriptional networks identifies TCF4 as a master regulator in schizophrenia. Sci Adv 2019;5:eaau4139.
31. Dries R, Stryjewska A, Coddens K, et al. Integrative and perturbation-based analysis of the transcriptional dynamics of TGFβ/BMP system components in transition from embryonic stem cells to neural progenitors. Stem Cells 2020;38:202-17.
32. Verschueren K, Remacle JE, Collart C, et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5’-CACCT sequences in candidate target genes. J Biol Chem 1999;274:20489-98.
33. Mowat DR, Croaker GD, Cass DT, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet 1998;35:617-23.
34. Cacheux V, Dastot-Le Moal F, Kääriäinen H, et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet 2001;10:1503-10.
35. Wakamatsu N, Yamada Y, Yamada K, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet 2001;27:369-70.
37. Mainardi P, Pastore G, Zweier C, Rauch A. Mowat-Wilson syndrome and mutation in the zinc finger homeo box 1B gene: a well defined clinical entity. J Med Genet 2004;41:e16.
38. Ishihara N, Yamada K, Yamada Y, et al. Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22-q24.1. J Med Genet 2004;41:387-93.
39. Yamada K, Yamada Y, Nomura N, et al. Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features. Am J Hum Genet 2001;69:1178-85.
40. Wilson M, Mowat D, Dastot-Le Moal F, et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am J Med Genet A 2003;119A:257-65.
41. Zweier C, Thiel CT, Dufke A, et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet 2005;48:97-111.
42. Zweier C, Horn D, Kraus C, Rauch A. Atypical ZFHX1B mutation associated with a mild Mowat-Wilson syndrome phenotype. Am J Med Genet A 2006;140:869-72.
43. Garavelli L, Zollino M, Mainardi PC, et al. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet A 2009;149A:417-26.
44. Ivanovski I, Djuric O, Caraffi SG, et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet Med 2018;20:965-75.
45. Garavelli L, Ivanovski I, Caraffi SG, et al. Neuroimaging findings in Mowat-Wilson syndrome: a study of 54 patients. Genet Med 2017;19:691-700.
46. Ricci E, Fetta A, Garavelli L, et al. Mowat Wilson Epilepsy Study Group. Further delineation and long-term evolution of electroclinical phenotype in Mowat Wilson Syndrome. A longitudinal study in 40 individuals. Epilepsy Behav 2021;124:108315.
47. Dastot-Le Moal F, Wilson M, Mowat D, et al. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat 2007;28:313-21.
48. Heinritz W, Zweier C, Froster UG, et al. A missense mutation in the ZFHX1B gene associated with an atypical Mowat-Wilson syndrome phenotype. Am J Med Genet A 2006;140:1223-7.
49. Ghoumid J, Drevillon L, Alavi-Naini SM, et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum Mol Genet 2013;22:2652-61.
50. Verstappen G, van Grunsven LA, Michiels C, et al. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum Mol Genet 2008;17:1175-83.
51. Wu LM, Wang J, Conidi A, et al. Zeb2 recruits HDAC-NuRD to inhibit notch and controls Schwann cell differentiation and remyelination. Nat Neurosci 2016;19:1060-72.
52. Birkhoff JC, Brouwer RWW, Kolovos P, et al. Targeted chromatin conformation analysis identifies novel distal neural enhancers of ZEB2 in pluripotent stem cell differentiation. Hum Mol Genet 2020;29:2535-50.
53. Huang X, Ferris ST, Kim S, et al. Differential usage of transcriptional repressor Zeb2 enhancers distinguishes adult and embryonic hematopoiesis. Immunity 2021;54:1417-1432.e7.
54. Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J 1978;14:182-4.
55. Amiel J, Rio M, de Pontual L, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet 2007;80:988-93.
56. Brockschmidt A, Todt U, Ryu S, et al. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet 2007;16:1488-94.
57. Zweier C, Peippo MM, Hoyer J, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am J Hum Genet 2007;80:994-1001.
58. Marangi G, Zollino M. Pitt-Hopkins Syndrome and Differential Diagnosis: a Molecular and Clinical Challenge. J Pediatr Genet 2015;4:168-76.
59. Whalen S, Héron D, Gaillon T, et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat 2012;33:64-72.
60. Fierro JA, Avina DA. Pitt-Hopkins syndrome: mental retardation, psychomotor and developmental delays with facial dysmorphism. J Pediatr Genet 2014;3:141-5.
61. Mary L, Piton A, Schaefer E, et al. Disease-causing variants in TCF4 are a frequent cause of intellectual disability: lessons from large-scale sequencing approaches in diagnosis. Eur J Hum Genet 2018;26:996-1006.
62. Goodspeed K, Newsom C, Morris MA, et al. Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. J Child Neurol 2018;33:233-44.
63. Remacle JE, Kraft H, Lerchner W, et al. New mode of DNA binding of multi-zinc finger transcription factors: deltaEF1 family members bind with two hands to two target sites. EMBO J 1999;18:5073-84.
64. Birkhoff JC, Korporaal AL, Brouwer RWW, et al. Zeb2 DNA-binding sites in ES cell derived neuroprogenitor cells reveal autoregulation and align with neurodevelopmental knockout mouse and disease phenotypes. bioRχiv 2021; doi: 10.1101/2021.07.06.451350.
65. Murre C, Mccaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MYOD, and MYC proteins. Cell 1989;56:777-83.
66. Conidi A, van den Berghe V, Leslie K, et al. Four amino acids within a tandem QxVx repeat in a predicted extended α-helix of the Smad-binding domain of Sip1 are necessary for binding to activated Smad proteins. PLoS One 2013;8:e76733.
67. Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5’ exon usage and splicing. PLoS One 2011;6:e22138.
68. Nurm K, Sepp M, Castany-Pladevall C, et al. Isoform-Specific Reduction of the Basic Helix-Loop-Helix Transcription Factor TCF4 Levels in Huntington’s Disease. eNeuro 2021;8:ENEURO.
69. Chen HY, Bohlen JF, Maher BJ. Molecular and cellular function of transcription factor 4 in Pitt-Hopkins syndrome. Dev Neurosci 2021;43:159-67.
70. Maduro V, Pusey BN, Cherukuri PF, et al. Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J Rare Dis 2016;11:62.
71. Bedeschi MF, Marangi G, Calvello MR, et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet 2017;60:565-71.
72. Forrest M, Chapman RM, Doyle AM, et al. Functional analysis of TCF4 missense mutations that cause Pitt-Hopkins syndrome. Hum Mutat 2012;33:1676-86.
73. Wang LH, Baker NE. E proteins and ID proteins: helix-loop-helix partners in development and disease. Dev Cell 2015;35:269-80.
74. Yang J, Horton JR, Li J, et al. Structural basis for preferential binding of human TCF4 to DNA containing 5-carboxylcytosine. Nucleic Acids Res 2019;47:8375-87.
75. Massari ME, Grant PA, Pray-grant MG, et al. A conserved motif present in a class of helix-loop-helix proteins activates transcription by direct recruitment of the SAGA complex. Molecular Cell 1999;4:63-73.
76. Bayly R, Chuen L, Currie RA, et al. E2A-PBX1 interacts directly with the KIX domain of CBP/p300 in the induction of proliferation in primary hematopoietic cells. J Biol Chem 2004;279:55362-71.
77. Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science 2004;305:1286-9.
78. Quevedo M, Meert L, Dekker MR, et al. Mediator complex interaction partners organize the transcriptional network that defines neural stem cells. Nat Commun 2019;10:2669.
79. van den Berghe V, Stappers E, Vandesande B, et al. Directed migration of cortical interneurons depends on the cell-autonomous action of Sip1. Neuron 2013;77:70-82.
80. Stryjewska A, Dries R, Pieters T, et al. Zeb2 regulates cell fate at the exit from epiblast state in mouse embryonic stem cells. Stem Cells 2017;35:611-25.
81. van Helden MJ, Goossens S, Daussy C, et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med 2015;212:2015-25.
82. Wang J, Farkas C, Benyoucef A, et al. Interplay between the EMT transcription factors ZEB1 and ZEB2 regulates hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity. PLoS Biol 2021;19:e3001394.
83. Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014;159:374-87.
84. Benito-Kwiecinski S, Giandomenico SL, Sutcliffe M, et al. An early cell shape transition drives evolutionary expansion of the human forebrain. Cell 2021;184:2084-2102.e19.
85. Kim H, Berens NC, Ochandarena NE, Philpot BD. Region and cell type distribution of TCF4 in the postnatal mouse brain. Front Neuroanat 2020;14:42.
86. Lennertz L, Rujescu D, Wagner M, et al. Novel schizophrenia risk gene TCF4 influences verbal learning and memory functioning in schizophrenia patients. Neuropsychobiology 2011;63:131-6.
87. Quednow BB, Ettinger U, Mössner R, et al. The schizophrenia risk allele C of the TCF4 rs9960767 polymorphism disrupts sensorimotor gating in schizophrenia spectrum and healthy volunteers. J Neurosci 2011;31:6684-91.
88. Jung M, Häberle BM, Tschaikowsky T, et al. Analysis of the expression pattern of the schizophrenia-risk and intellectual disability gene TCF4 in the developing and adult brain suggests a role in development and plasticity of cortical and hippocampal neurons. Mol Autism 2018;9:20.
89. Sripathy SR, Wang Y, Moses RL, et al. Generation of 10 patient-specific induced pluripotent stem cells (iPSCs) to model Pitt-Hopkins syndrome. Stem Cell Res 2020;48:102001.
90. Gheldof A, Hulpiau P, van Roy F, De Craene B, Berx G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci 2012;69:2527-41.
91. Hegarty SV, Sullivan AM, O’Keeffe GW. Zeb2: a multifunctional regulator of nervous system development. Prog Neurobiol 2015;132:81-95.
92. Epifanova E, Babaev A, Newman AG, Tarabykin V. Role of Zeb2/Sip1 in neuronal development. Brain Res 2019;1705:24-31.
93. Scott CL, Omilusik KD. ZEBs: novel players in immune cell development and function. Trends Immunol 2019;40:431-46.
94. Higashi Y, Maruhashi M, Nelles L, et al. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 2002;32:82-4.
95. Seuntjens E, Nityanandam A, Miquelajauregui A, et al. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat Neurosci 2009;12:1373-80.
96. McKinsey GL, Lindtner S, Trzcinski B, et al. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013;77:83-98.
97. Deryckere A, Stappers E, Dries R, et al. Multifaceted actions of Zeb2 in postnatal neurogenesis from the ventricular-subventricular zone to the olfactory bulb. Development 2020;147:dev184861.
98. Vivinetto AL, Kim ID, Goldberg DC, et al. Zeb2 is a regulator of astrogliosis and functional recovery after CNS injury. Cell Rep 2020;31:107834.
99. Quintes S, Brinkmann BG, Ebert M, et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat Neurosci 2016;19:1050-9.
100. Boland BS, He Z, Tsai MS, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol 2020;5:eabb4432.
101. Gladka MM, Kohela A, Molenaar B, et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat Commun 2021;12:84.
102. de Haan W, Dheedene W, Apelt K, et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc Res 2022;118:1262-75.
103. Vandamme N, Denecker G, Bruneel K, et al. The EMT transcription factor ZEB2 promotes proliferation of primary and metastatic melanoma while suppressing an invasive, mesenchymal-like phenotype. Cancer Res 2020;80:2983-95.
104. Turovskaya MV, Babaev AA, Zinchenko VP, et al. Sip-1 mutations cause disturbances in the activity of NMDA- and AMPA-, but not kainate receptors of neurons in the cerebral cortex. Neurosci Lett 2017;650:180-6.
105. Jeub M, Emrich M, Pradier B, et al. The transcription factor Smad-interacting protein 1 controls pain sensitivity via modulation of DRG neuron excitability. Pain 2011;152:2384-98.
106. Pradier B, Jeub M, Markert A, et al. Smad-interacting protein 1 affects acute and tonic, but not chronic pain. Eur J Pain 2014;18:249-57.
107. Khan RAW, Chen J, Wang M, et al. A new risk locus in the ZEB2 gene for schizophrenia in the Han Chinese population. Prog Neuropsychopharmacol Biol Psychiatry 2016;66:97-103.
108. Takagi T, Nishizaki Y, Matsui F, Wakamatsu N, Higashi Y. De novo inbred heterozygous Zeb2/Sip1 mutant mice uniquely generated by germ-line conditional knockout exhibit craniofacial, callosal and behavioral defects associated with Mowat-Wilson syndrome. Hum Mol Genet 2015;24:6390-402.
109. Rannals MD, Maher BJ. Molecular mechanisms of transcription factor 4 in Pitt hopkins syndrome. Curr Genet Med Rep 2017;5:1-7.
110. Braun K, Häberle BM, Wittmann MT, Lie DC. Enriched environment ameliorates adult hippocampal neurogenesis deficits in Tcf4 haploinsufficient mice. BMC Neurosci 2020;21:50.
111. Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci 2000;1:191-8.
112. Kennedy AJ, Rahn EJ, Paulukaitis BS, et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep 2016;16:2666-85.
113. Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet 2012;21:2873-88.
114. Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A 2007;104:15382-7.
115. Fischer B, Azim K, Hurtado-Chong A, et al. E-proteins orchestrate the progression of neural stem cell differentiation in the postnatal forebrain. Neural Dev 2014;9:23.
116. Li H, Zhu Y, Morozov YM, et al. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol Psychiatry 2019;24:1235-46.
117. Mesman S, Bakker R, Smidt MP. Tcf4 is required for correct brain development during embryogenesis. Mol Cell Neurosci 2020;106:103502.
118. Thaxton C, Kloth AD, Clark EP, et al. Common pathophysiology in multiple mouse models of Pitt-Hopkins syndrome. J Neurosci 2018;38:918-36.
119. Schoof M, Hellwig M, Harrison L, et al. The basic helix-loop-helix transcription factor TCF4 impacts brain architecture as well as neuronal morphology and differentiation. Eur J Neurosci 2020;51:2219-35.
120. Phan BN, Bohlen JF, Davis BA, et al. A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci 2020;23:375-85.
121. Weng Q, Chen Y, Wang H, et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012;73:713-28.
122. Zhang Y, Cai Y, Wang Y, et al. Survival control of oligodendrocyte progenitor cells requires the transcription factor 4 during olfactory bulb development. Cell Death Dis 2021;12:91.
123. Cisse B, Caton ML, Lehner M, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008;135:37-48.
124. Hill MJ, Killick R, Navarrete K, et al. Knockdown of the schizophrenia susceptibility gene. TCF4 ;42:181-8.
125. Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One 2013;8:e73169.
126. Chen T, Wu Q, Zhang Y, et al. Tcf4 controls neuronal migration of the cerebral cortex through regulation of bmp7. Front Mol Neurosci 2016;9:94.
128. Kloet SL, Baymaz HI, Makowski M, et al. Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J 2015;282:1774-85.
129. Torchy MP, Hamiche A, Klaholz BP. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol Life Sci 2015;72:2491-507.
130. Fröb F, Wegner M. The role of chromatin remodeling complexes in Schwann cell development. Glia 2020;68:1596-603.
131. D’Souza L, Channakkar AS, Muralidharan B. Chromatin remodelling complexes in cerebral cortex development and neurodevelopmental disorders. Neurochem Int 2021;147:105055.
132. Benayoun BA, Pollina EA, Ucar D, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014;158:673-88.
133. Chen K, Chen Z, Wu D, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet 2015;47:1149-57.
134. Miquelajauregui A, Van de Putte T, Polyakov A, et al. Smad-interacting protein-1 (Zfhx1b) acts upstream of Wnt signaling in the mouse hippocampus and controls its formation. Proc Natl Acad Sci U S A 2007;104:12919-24.
135. Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease-can we wrap it up? Brain 2011;134:1882-900.
136. de Putte T, Francis A, Nelles L, van Grunsven LA, Huylebroeck D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum Mol Genet 2007;16:1423-36.
137. Szu J, Wojcinski A, Jiang P, Kesari S. Impact of the Olig Family on Neurodevelopmental Disorders. Front Neurosci 2021;15:659601.
138. Hennig KM, Fass DM, Zhao WN, et al. WNT/β-catenin pathway and epigenetic mechanisms regulate the pitt-hopkins syndrome and schizophrenia risk gene. TCF4 ;3:53-71.
139. Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP. Dynamic EMT: a multi-tool for tumor progression. EMBO J 2021;40:e108647.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Meert L, Birkhoff JC, Conidi A, Poot RA, Huylebroeck D. Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome) . Rare Dis Orphan Drugs J 2022;1:8. http://dx.doi.org/10.20517/rdodj.2022.03
AMA Style
Meert L, Birkhoff JC, Conidi A, Poot RA, Huylebroeck D. Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome) . Rare Disease and Orphan Drugs Journal. 2022; 1(2): 8. http://dx.doi.org/10.20517/rdodj.2022.03
Chicago/Turabian Style
Meert, Lize, Judith C. Birkhoff, Andrea Conidi, Raymond A. Poot, Danny Huylebroeck. 2022. "Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome) " Rare Disease and Orphan Drugs Journal. 1, no.2: 8. http://dx.doi.org/10.20517/rdodj.2022.03
ACS Style
Meert, L.; Birkhoff JC.; Conidi A.; Poot RA.; Huylebroeck D. Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome) . Rare. Dis. Orphan. Drugs. J. 2022, 1, 8. http://dx.doi.org/10.20517/rdodj.2022.03
About This Article
Copyright

Data & Comments
Data


 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 29
likes
Like This Article 29
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.