
Rare diseases: specific challenges for sustainable accessibility of treatments for patients
 0
0Abstract
In late 2021, the health technology assessment of the French National Authority for Health was seized by the French Ministry of Solidarity and Health to address a specific challenge, the identification of solutions contributing to the development of the methodological expertise in new types of clinical studies for rare diseases. Experts from the rare diseases environment were gathered by OrphanDev, the French network of expertise dedicated to rare diseases. They allowed to identify some of the current issues in France concerning rare diseases, and then present different solutions, in particular related to the evaluation process of orphan drugs and the collection of data on rare diseases.
Keywords
INTRODUCTION
Since 1990, France has been committed to elaborating and implementing public health policies dedicated to rare diseases. This commitment notably led to the creation of three national plans for rare diseases [namely National Rare Diseases Plan 1 (PNMR1), PNMR2, and PNMR3]. The specific features of France arising from these policies, and the pre-existing features involved in rare diseases are presented hereafter in Table 1.
French features involved in rare diseases
| French features | Acronyms | Definition | Designation in the text |
| Agence nationale de sécurité du médicament et des produits de santé | ANSM | Public body allows, on behalf of the State, access to health products in France and ensures their safety throughout their life cycle. It promotes access to innovative products via authorization procedures adapted to each stage of the life of the medicinal product before and after it is placed on the market Through assessments, expertise, and surveillance policy, the health products are ensured to be available in France, safe, effective, accessible, and well used | The French Agency for the Safety of Health Products |
| Amélioration du service médical rendu&Service médical rendu | ASMR&SMR | Two opinions issued by the HAS CT were used as recommendations for the reimbursement of the drug and its rate of reimbursement (SMR) and to contribute to the negotiation of the drug price (ASMR) Evaluation of the clinical added value of a drug Five levels of ASMR have been defined: - ASMR I: Major therapeutic progress - ASMR II: Significant improvement in terms of therapeutic efficacy and/or reduction of adverse events - ASMR III: Modest improvement in terms of therapeutic efficacy and/or reduction of adverse events - ASMR IV: Minor improvement in efficiency and/or utility - ASMR V: No therapeutic progress Criterion takes into account the seriousness of the pathology and the data specific to the drug in a given indication Three levels of SMR have been defined: - Major or important SMR (clinical benefit) - Moderate or low SMR, justifying, however, the reimbursement - Insufficient SMR to justify coverage by the community | Clinical added value Actual clinical benefit |
| Banque Nationale de Données Maladies Rares | BNDMR | French national rare diseases data bank that aims to provide France with a homogeneous collection of data based on a minimum data set to document the management and health status of rare disease patients in French expert centers, and to better evaluate the effect of national plans | National Rare Diseases Data Bank |
| Centres de Références de Maladies Rares | CRMR | The CRMR are specialized in one or more rare diseases and put their expertise into action in the fields of care, teaching and research | Centers of reference for rare diseases |
| Comité de transparence | CT | Scientific body composed of doctors, pharmacists, specialists in methodology and epidemiology. It evaluates medicines that have obtained their marketing authorization (MA), when the laboratory that markets them wishes to obtain their inclusion on the list of reimbursable medicines | Transparency Committee |
| Filières de santé maladies rares | FSMR | The 23 “filières de santé” constitute a network where each of them covers a broad and coherent field of diseases. They coordinate the collective action of the CRMR, privates, and healthcare professionals for rare diseases. Their aim is to improve care, research, education, training, and information. | Rare Diseases Healthcare Network (RDHN) |
| Haute Autorité de Santé | HAS | The HAS is an independent public authority of a scientific nature. Focusing on the benefits for the population, it aims to develop quality in the health, social, and medico-social fields The HAS informs public authorities, optimizes the practices and organizations of healthcare professionals and strengthens the choice of the population The HAS (1) evaluates drugs, medical devices, and procedures for reimbursement (2) recommends good practices and elaborates public health recommendations, and (3) measures and improves the quality of hospitals, clinics, community medicine, and social and medico-social establishments | French National Authority for Health |
| Programmes Nationaux de Diagnostic et de Soins | PNDS | Best practice guidelines for rare diseases. The objective is to explain to the professionals concerned the optimal diagnostic and therapeutic management and care pathway of a patient suffering from a given rare disease | National Diagnosis and treatment protocol |
| Protocole d’Utilisation Thérapeutique et de Recueil de Données | PUT-RD | Reference document for the healthcare industry which compiles all the data collected in the framework of early access | Therapeutic use protocol and compilation of information |
| Set de données minimum traitement | SDM-T | Document created for each patient suffering from a rare disease and received in a CRMR. It constitutes the common base of information for all rare diseases and for all those involved in the care of the patient | Minimum Treatment Data Set |
| Système national des données de santé | SNDS | A National Healthcare System Database that brings together the main existing public health databases (health insurance, health facilities, complementary health insurance, public housing for persons with disabilities). It aims to improve knowledge about medical care and broaden the scope of research, studies, and evaluations in the health field | French National Healthcare System Database |
However, despite these years of innovations in rare diseases, there are still various challenges that need to be addressed. In that context, in late 2021, the Health Technology Assessment of the French National Authority for Health (HAS) was seized by the French Ministry of Solidarity and Health. Different actors in the rare diseases field were required to address a common challenge, the identification of solutions contributing to the development of the methodological expertise in new types of clinical studies for rare diseases.
One of the experts approached was OrphanDev, the French network of expertise dedicated to rare diseases funded by F-CRIN, and specialized in supporting researchers, clinicians, and the health industry actors in the development of drugs for rare diseases. OrphanDev gathered several experts from academic teams, pharmaceutical industries, authorities, and Rare Diseases Healthcare Network (RDHN). The discussions allowed, in the first place, to have an overview of the existing issues in France concerning rare diseases, and in the second place, to agree on the proposals that should be made. We present, hereafter, the different solutions identified that represent an improvement for rare diseases, especially concerning the evaluation process, the implication for patients and experts in this process, and finally, the collection, accessibility, and use of the data on rare diseases.
A SPECIFIC EVALUATION PATHWAY FOR TREATMENTS IN RARE DISEASES
In France, there is no specific evaluation pathway dedicated to orphan drugs. This implies that orphan drugs are evaluated according to the same evaluation process as common drugs. This evaluation process is presented in Figure 1.
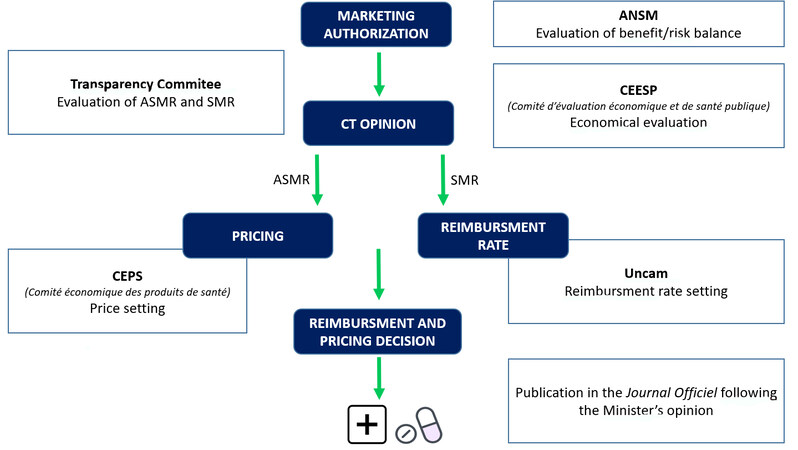
Figure 1. Access to the market of a drug in France: from marketing authorization to reimbursement and pricing decisions.
This absence of a specific evaluation process for orphan drugs results in a significant time to availability (660 days between marketing authorization and the date of availability[1], which is notably impacted by the regulatory instruction period of the HAS that is set to 90 days (These delays do not include the possibility for orphan drugs to have early access: the regulatory instruction period for early access is set to 60days, and the drug is immediately accessible to the patients).
These are the reasons why there is a consensus on the need to evolve the current evaluation procedure for rare disease treatments, especially to accelerate their availability.
Take into account the specificities of orphan drugs during the evaluation process
Creation of an ad hoc procedure
A systematic consideration of the specificities of orphan drugs requires the establishment of a dedicated evaluation procedure for rare diseases. Within the HAS, this ad hoc rare diseases procedure could be managed by dedicated interlocutors, following the example of the EMA’s Committee for Orphan Medicinal Products, with the intervention of rapporteurs specialized in rare diseases and, if necessary, in pediatrics.
For products that benefit from early access, it should be possible to postpone the final evaluation by maintaining derogatory medical care. Indeed, in some cases, the time required to collect data from clinical studies or during the early access phase does not allow to fully document the impact of a drug, particularly for drugs treating slowly progressing rare diseases. Among these, for rare, serious, and genetic diseases, several years must be waited to observe the effect of a treatment on a large population of patients. This conflicts with the doctrine of the Transparency Commission (CT), which wishes to have proof of the impact of the drug on patient morbidity and mortality as soon as the marketing authorization is granted[2].
Agreeing to postpone the final evaluation, while allowing controlled and regulated use within the framework of early access, represents an opportunity for the HAS to have more mature data and sufficient hindsight on the data collected to allow an informed evaluation.
Take into account the status of “Orphan Medical Product” and its “significant benefit” recognized by the EMA and the European Commission in the evaluation
Application of the European regulation
The European regulation (EC 141/2000), which gives the definition of an orphan drug in Europe, recognizes de facto the notion of “significant benefit” for orphan drugs. This significant benefit indicates that an orphan drug improves the treatment outcomes compared to existing solutions. Compliance with this provision in the French evaluation process implies changes in the qualification of scores related to this notion of benefit:
- The actual clinical benefit (SMR) of orphan drugs should be considered important, based on the medical needs expressed by the experts and the “significant benefit” of the European MA.
- There should be a creation of an “orphan” ASMR, corresponding to the fact that the drug brings a benefit to the patients as stated in the European regulation.
- The orphan ASMR could be transitory, in the event of uncertainties that could be resolved in the short or medium term, and would be disconnected from the classic system at the time of the initial evaluation; it would be reclassified as an ASMR I, II, III or IV [Table 1] either after the resolution of the uncertainties or after 3 years.
- The expected place of orphan drugs in the therapeutic strategy will first be determined and then accessed by the experts.
Recognizing this notion of “significant benefit” for orphan drugs during the French evaluation process will eventually allow the drug (1) to reach more quickly the CT opinion publication; (2) to be easily accessible and reimbursed for patients with a list price consistent with other prices published in Western European countries; (3) to have a specific regulated price; and (4) to be accessible to patients on a long term continuous basis.
Mobilize and engage all stakeholders, patients, and their representatives, RDHN, pharmaceutical companies, and health authorities through a multipartite commitment during the evaluation
This commitment could be linked to the reform of the early access and could anticipate a re-evaluation of available clinical data at a predefined date by the authorities, complementary to the data collected in the context of early access.
Collective exchange and commitment
The involvement of all stakeholders in a transparent dialogue over time is essential. It would bring together the evaluation bodies (HAS Transparency Commission, ANSM), the laboratories, the patient representatives, RDHN, and the BNDMR.
The first exchanges would take place before the conditional Rare Disease procedure, not exceeding more than 6 months. It will allow the joint definition of the conditions of feasibility and the means to be implemented to ensure compliance with the deadlines for making data available and the level of completeness of the relevant data. It takes into account and is consistent with all the pre-existing tools and the requests from the EMA. This would be materialized by a multipartite contract of all the stakeholders, describing:
- The modalities of data collection and evaluation, the criteria, stages, conditions, and expected elements.
- The duration of the conditionality of the initial evaluation. This duration must take into account the natural evolution of the disease in question and allow the collection of relevant data for the evaluation. In any case, a deadline is set collectively for the analysis and the final evaluation, at most 3 years after the MA.
- The schedule for monitoring the progress of data collection.
- The measures applied in case of non-compliance (corrective actions, penalties, etc.).
Implement a real-life evaluation to supplement the data from “classic” clinical trials and integrate flexibility in the evaluation processes
The criteria evaluated today by the HAS CT mainly concern the quantity of additional effect and the clinical relevance of the drug in relation to its comparator, generally demonstrated in terms of the impact of the primary endpoint on morbidity, mortality, and quality of life. The slow evolution of rare diseases and their heterogeneity makes it difficult, if not impossible, to set up clinical studies demonstrating the impact of treatments on morbidity and mortality within a specific timeframe[3].
Data collection for each diseases
The multipartite contract would provide for the modalities and supports for the collection of the expected data: use, creation, or development of a registry specific to the, such as a natural history study (collection of information on the disease, the impact on patients’ lives, and the medical resources used) or use of the minimum treatment data set (SDM-T) of the BNDMR.
The cost of this data collection would be funded, fully or partly, by the pharmaceutical/biotech company.
The multipartite contract also specifies the quality constraints and the respective obligations and responsibilities of the actors involved, particularly in terms of data completeness and proper use as well as quality control.
Validation of the protocol and data collection methods by the authorities
Beforehand, the authorities will have established and shared with the actors a guide specific to rare diseases specifying the data and the methods of their collection as well as the methodology of the accepted statistical analyses.
The manufacturers submit a protocol for therapeutic use and data collection established in partnership with experts of the pathology, including patients, and validated by the HAS either for early access or during the primary evaluation for products benefiting from an ASMR “orphan”.
Validation of intermediate criteria and specific scales
In the multipartite contract, the method of analysis of the clinical data collected will be discussed, particularly with regard to criteria including intermediate, non-hierarchical secondary, and quality of life presented in the initial studies. This analysis may allow the validation of specific scales (results, relevant clinical validity thresholds) that are essential for the evaluation of therapeutic innovations.
Minimum Data Set - Treatments of the BNDMR
This additional data collection module could be deployed in the centers and RDHN considered, according to the ad-hoc need (early access or compassionate, real-life study promoted by a laboratory).
The items of this module could include the dates of visits, the name of the drug or medical device used, the prescription context, the treatment modalities, the route of administration, the PROM and efficacy data, the adverse effects observed, and so on.
During the evaluation of a drug by the authorities, because PROM and efficacy data are the two items of interest, they could be adapted and developed “on demand” according to the situation.
THE BENEFIT OF A SUI GENERIS CLINICAL EXPERTISE FOR RARE AND ULTRA-RARE DISEASES: THE CONTRIBUTION OF RDHN
The French structuration of the healthcare system for patients suffering from rare diseases through the 23 RDHN is an element that must be capitalized on in the process of evaluating treatments. Soliciting the CRMRs via the RDHN would make it possible to obtain specialized scientific and clinical expertise on the pathology concerned. This element is essential given the incidence and prevalence of the diseases concerned.
This request is initiated by the HAS and must be made upstream.
Collegial expert opinion
The organization of the RDHN allows a collegial contribution through its Medicines Commission (or if necessary, by setting up a small ad hoc group of 2-3 experts, for example, those who participated in the drafting of the PNDS).
Assessment of needs and place in the therapeutic strategy:
The expected content of this expertise is
- To review the met/partially met/unmet needs of the disease
- To associate a pediatric opinion if necessary
- To clarify the comparators:
● The health insurance standard of care
● The reality in the field and the use outside the MA
● French specificities, if any
- To define and validate the evaluation criteria
- In a second phase, after the new product has been made available, to evaluate its place in the therapeutic strategy
Co-construction of data collection
The RDHN participate in the creation (or development) of registries and databases for each disease with the other actors concerned, pharmaceutical/biopharmaceutical companies, the BNDMR, and European databases when appropriate, and provide them with exhaustive information with regard to the data collected.
These data are intended to resolve uncertainties identified at the beginning of the evaluation or collect real-life data when it is not possible to set up a randomized Phase III study and increase the level of knowledge regarding clinical efficacy, product safety, and cost-effectiveness of treatments.
European level for ultra-rare diseases
When the number of patients is very small and data are scarce, it would be optimal to involve, as is the case in other European countries, the established structures collecting data on the disease at a European level (ERNs or other European registers).
PARTICULAR ATTENTION TO THE SOLICITATION OF PATIENTS
The participation of patients in the evaluation process encounters several difficulties in terms of time and resources, and on the question of representativeness[4-6]:
- As part of the HAS evaluation of health products, patients’ associations can submit a contribution using a questionnaire to collect patients’ views. However, the tight deadlines for submission (45 days), the structuring of patient associations in rare diseases composed of volunteers, and the high technicality of these questionnaires do not always allow an effective contribution.
- When surveys are produced by associations, they are often considered biased because the responders are essential members of patient associations and are not always considered representative of the general population affected by the disease in question.
In addition, the inclusion of contributions from patient representatives varies. During the evaluation, these contributions are not systematically considered and integrated in the same way as the clinical indicators, which does not encourage patients to make time to respond.
The challenge for patient organizations is to provide objective and usable data for the health authorities and thus learn to speak “the same language” in order to be understood and valued. Supporting patient associations in writing their contributions would strengthen them and thus enable them to better meet the methodological requirements of the HAS[4].
Participation in the discussion
The participation of the patient representatives in the peer review would allow them to be involved from the beginning and at all stages of the evaluation process. Thus, they will be able to better understand the expectations of the authorities, have more time to analyze and express the patients’ needs, and clarify the reflection on data collection and definition of the criteria taken into account.
THE CONTRIBUTION OF REAL-LIFE DATA: RECOGNITION AND QUALITY ISSUES
The development of registries and databases for each disease is an essential element for improving knowledge of the diseases and their management.
In the context of the development of new therapies, the co-construction of data collection (registry, adaptation of an existing registry, database, SDM-T) by all stakeholders and with the authorities, would represent an opportunity to have an optimized tool.
The objective of this tool is to avoid duplicated and redundant databases, to guide defining the expected data and collection methods, as well as the methodology of the accepted statistical analyses, to make their financing permanent and secure, to improve the relevance and the possibilities of analysis of the collected data and, above all, to be able to contribute to the recognition of the significant benefit brought by the evaluated drug.
The specifications for data collection must define the rules of ownership and use of the data, the quality and independence requirements, the criteria for completeness and the distribution of tasks and responsibilities between actors. Therefore, the tool relies on four major strengths:
- Quality. The relevance of data and analysis is improved by the co-construction and the compliance of the guide provided by the authorities: definition of criteria by the experts of the field, with the patients’ input.
- Independence. The data is carried by the academic community (responsibility for the data lies with the RDHN that generates and uses it). The laboratories interested in the interpretation of the data would only have access to the aggregated data.
- Homogeneity. The interest is to have a single, interoperable database with a single format useful at least at the European level by pathology, rather than several databases.
- Centralization. The objective, a fortiori for ultra-rare diseases, is to be able to develop the tool on a European scale.
Interactive system and patient participation
Depending on the pathologies and feasibility, it could include an interactive system of data entry directly by the patients and/or caregivers with medical supervision. It could be based on the digital surveys used in pharmacovigilance for the collection of side effects and the monitoring of relevant data.
Linkage with the SNDS and the role of the BNDMR or existing rare disease registries
The co-construction with the BNDMR aims to have a complete tool, allowing to link clinical data, data transmitted by the patients (PROs, PROMs), and medico-administrative data available in France through the SNDS/Health Data Hub.
The treatment module of the BNDMR could be coupled with SNDS data for a more detailed analysis of the care pathway.
Depending on the situation, the BNDMR could also supply existing registries with the data it has, depending on the completeness of the tool.
Analysis and use of data
In addition to the guidelines provided by the guide on real-life studies for the evaluation of medicines written by the HAS, the stakeholders, authorities, and data collection actors will define and validate the methodology applied for the analysis and valorization of the real-life data collected.
SYNTHESIS OF THE SOLUTIONS PROPOSED
According to the experts in the field of rare diseases gathered by OrphanDev, answering this challenge of identification of solutions contributing to the improvement of rare diseases in France requires the application of the different points summarized hereafter.
- There must be a systematic consideration of the orphan drug status according to the European regulation by introducing a temporary “Orphan drug” ASMR equivalent to an ASMR IV and to a significant SMR.
- Dedicated interlocutors and specialized rare disease/pediatric reporters must be involved in the clinical and medico-economic evaluation.
- A specific rare diseases evaluation pathway for products with early access must be created, in which the final evaluation can be postponed in time.
- A timeline for collective discussions (envisaged to last a maximum of 6 months) and a multipartite engagement of all stakeholders and authorities should be established. The objective is to agree on feasibility conditions, the means allowing to respect the deadlines for making the data available, and completeness level of the relevant data, in line with all the pre-existing tools and the requests of the EMA.
- The evaluation time that takes into account the generation and analysis of the data foreseen in the multipartite commitment should be extended. Annual defined steps and a deadline of 3 years should be considered.
- A collegial and specialized expertise via the drug commissions of the rare diseases and/or data collection structures at the European level must be set up. It will define the place of the drugs in the therapeutic strategy and define and validate the evaluation criteria.
- Patients’ representatives must be integrated upstream and at all stages of the process.
- A data collection by disease (registry, database) must be developed. Linked to the SNDS, it will be able to analyze real-life data.
- A clear and readable methodology for the analysis and use of the data collected must be proposed.
DECLARATIONS
Acknowledgments
OrphanDev (https://www.orphan-dev.org/) is a French national network funded by F-CRIN aiming to bring solutions to patients suffering from rare diseases.
Author’s contributions
Discussion and writing of the manuscript: Cortial L, Boyer PO, Forget S, Le Joubioux R, Blin O, Mouthon F
Availability of data and materials
Not applicable.
Financial support and sponsorship
Not applicable .
Conflict of interest
All authors declared that there are no conflict of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
REFERENCES
1. Newton M, Scott K, Troein P. EFPIA patients W.A.I.T. indicator 2021 survey. Available from: https://www.efpia.eu/media/636821/efpia-patients-wait-indicator-final.pdf [Last accessed on 6 Jun 2022].
2. HAS. Transparency committee doctrine. Available from: https://www.has-sante.fr/upload/docs/application/pdf/2019-07/doctrine_de_la_commission_de_la_transparence_-_version_anglaise.pdf [Last accessed on 6 Jun 2022].
3. HAS. Real-world studies for the assessment of medicinal products and medical devices. Available from: https://www.has-sante.fr/upload/docs/application/pdf/2021-10/real-world_studies_for_the_assessment_of_medicinal_products_and_medical_devices.pdf [Last accessed on 6 Jun 2022].
4. PFIZER. Plaidoyer pour une reconnaissance de la perspective patient dans l’évaluation des prodiits de sante. Available from: https://www.pfizer.fr/sites/default/files/inline-files/Plaidoyer%20pour%20la%20reconnaissance%20de%20la%20perspective%20patient%20dans%20l%27%C3%A9valuationVF.pdf [Last accessed on 6 Jun 2022].
5. Gesbert C, André-Vert J, Guerrier M, et al. The contribution of French patient and consumer groups to health technology assessments over a 2-year period: an observational retrospective study. Int J Technol Assess Health Care 2021;37:e48.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cortial L, Boyer PO, Forget S, Le Joubioux R, Blin O, Mouthon F. Rare diseases: specific challenges for sustainable accessibility of treatments for patients. Rare Dis Orphan Drugs J 2022;1:12. http://dx.doi.org/10.20517/rdodj.2022.08
AMA Style
Cortial L, Boyer PO, Forget S, Le Joubioux R, Blin O, Mouthon F. Rare diseases: specific challenges for sustainable accessibility of treatments for patients. Rare Disease and Orphan Drugs Journal. 2022; 1(3): 12. http://dx.doi.org/10.20517/rdodj.2022.08
Chicago/Turabian Style
Cortial, Lucas, Pierre-Olivier Boyer, Sylvain Forget, Ronan Le Joubioux, Olivier Blin, Franck Mouthon. 2022. "Rare diseases: specific challenges for sustainable accessibility of treatments for patients" Rare Disease and Orphan Drugs Journal. 1, no.3: 12. http://dx.doi.org/10.20517/rdodj.2022.08
ACS Style
Cortial, L.; Boyer P.O.; Forget S.; Le Joubioux R.; Blin O.; Mouthon F. Rare diseases: specific challenges for sustainable accessibility of treatments for patients. Rare. Dis. Orphan. Drugs. J. 2022, 1, 12. http://dx.doi.org/10.20517/rdodj.2022.08
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 18 clicks
Cite This Article 18 clicks

 Like This Article 7
likes
Like This Article 7
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.