
Targeting neutrophil serine proteinases in alpha-1 antitrypsin deficiency
 0
0
Abstract
Alpha-1 antitrypsin (AAT) is the most abundant irreversible serine proteinase inhibitor in the circulation and plays a major role in protecting lung tissue against destruction from neutrophil serine proteinases. Genetic mutation of AAT leads to reduced circulating levels and AAT deficiency (AATD) which is associated with an increased risk of developing emphysema. This observation suggests that the balance between AAT and neutrophil serine proteinase is crucial in maintaining tissue homoeostasis. In AATD, the overexuberant proteinase activity resulting from inadequate AAT control creates a self-perpetuating inflammatory cycle, driving progressive tissue injury. Re-establishing this physiological balance is therefore critical for preserving lung architecture, function, and abrogating disease progression.
Several avenues within this pathophysiological pathway are being explored. This chapter addresses the pathophysiological process, current treatments targeting the pathway, and alternative approaches within the pathway that can potentially mitigate proteinase imbalance.
Keywords
INTRODUCTION
Alpha-1 antitrypsin [(AAT); also known as Serine Protease Inhibitor (SERPIN) family A1] is a polymorphic protein expressed in a codominant fashion by 2 alleles on chromosome 14[1]. It has a modulatory role in inflammation showing an acute phase response in its manufacture and release from the liver (the major source of production) and increased diffusion into the affected tissues/organs where it plays a primary role in protecting tissues from the destructive effect of serine proteinases.
The importance of this role is indicated by the genetic deficiency first observed in the 1960s[2]. Of the initial 5 cases identified, 3 had severe early onset basal emphysema and subsequent family studies confirmed the inheritance of the deficiency and the pulmonary association that appeared both spontaneously (with no recognised external risk factors[3]) but was accelerated by cigarette smoking[4]. Although other clinical associations such as panniculitis, systemic vasculitis, and liver cirrhosis are also recognised associations, it is the pulmonary disease that dominates.
Much has been learnt about the pathophysiology of the disease since the early observation of plasma deficiency. Over 150 variants of the protein have been identified, although most are associated with apparently “normal” concentrations and function. Deficiency can vary from partial due to heterozygosity of deficient genes with a normal gene to complete absence of AAT related mostly to point mutations and frameshift truncating mutations, although these are especially rare[5]. The majority of clinically relevant deficient cases are those homozygous for the Z variant which has a point mutation resulting in an amino acid substitution at position 342 (Glu to Lys) in the mature protein. This substitution can be identified through the genomic databases as rs28929474-p.Glu366Lys. As a result, this produces a protein that has a reduced association rate with its target enzymes[6-8]. This mutation also increases the likelihood of the protein to polymerise, particularly in the liver, impeding its secretion (by approximately 80%), resulting in plasma concentrations of ~5 µM[6-8].
The most relevant targets for AAT are serine proteinases and especially those stored in and released by circulating and migrating neutrophils. In the 1970s, the first report on the pulmonary damaging effect of neutrophil elastase was published[9]. Although this enzyme retains its importance as the direct mediator of emphysema and damage to many vital immune functions in the lung, it should be noted that two other enzymes stored in the same granules in the neutrophil (Proteinase 3 and Cathepsin G) can play similar damaging roles.
In the 1980s, the link between AAT deficiency (AATD) and emphysema was well established and the logical way to manage the condition was to enhance the circulating and hence lung concentration of AAT. Purification of plasma AAT from blood donors proved feasible and intravenous administration raised both the plasma[10] and lung[11] concentrations of AAT to safe and protective levels. Based on these studies, the United States Food and Drug Administration (FDA) approved weekly intravenous augmentation therapy for Z homozygous individuals on biochemical efficacy alone.
Because AATD is relatively rare (1 in 1500 to 1 in 5000) related to migration patterns from the Baltic areas[12], it was not felt feasible to carry out classical placebo-controlled studies to demonstrate clinical efficacy on the physiological progression of emphysema. However, observational studies[13,14] suggested a benefit. With the advent of highly sensitive quantitative computed tomographic density measurements of the lung as a direct measure of the emphysema process, the powering of studies to demonstrate the efficacy of augmentation therapy became feasible. Initial observational[13,14] and placebo-controlled studies[15-17] and a subsequent adequately powered placebo-controlled study[18] confirmed the ability of intravenous AAT to slow the progression of emphysema.
However, intravenous augmentation therapy is both expensive and inconvenient for the patient and remains unavailable in many countries. Research has continued into the pathophysiology of chronic lung disease including the processes relevant to AATD that suggest alternative strategies may be equally or potentially more effective in correcting the imbalance that occurs when enzymes such as elastase are released into an environment where deficient AAT fails to adequately control the enzyme activity exceeding its physiological role and leading to excessive tissue damage. The mechanism and potential strategies to enhance protection in AATD are discussed below.
The pathophysiology of emphysema
The proteinase/antiproteinase balance theory
AAT is the most abundant serine proteinase inhibitor in the circulation and plays a major role in the lung by largely entering the tissues via simple transudation and therefore increases in the presence of local inflammation[19]. In healthy individuals, plasma concentration of AAT ranges between 20 to 40 µM resulting in interstitial concentrations of roughly 80% of that in plasma. The other significant lung inhibitor is the secretory leukocyte proteinase inhibitor (SLPI) produced by mucous glands and bronchoepithelial cells and can be secreted basally into the interstitium[20]. Although a reversible inhibitor (unlike AAT, which is irreversible), it is better at protecting elastin from neutrophil elastase than AAT[21] despite being unable to inhibit Proteinase 3[22].
When the physiological balance between these enzymes (as in AATD) is disturbed, excessive tissue breakdown occurs as the proteinases will have a longer duration and radius of activity[23,24]. During neutrophilic inflammation, the neutrophil traffic and elastase load to the lung is increased. In those with normal AAT, the acute phase response increases AAT production and inflammation increases its penetration into the lung leading to modulation of inflammation. In AATD, the AAT concentration is too low at baseline and inflammation is greater than in patients with normal AAT which is associated with a poor acute phase response[25]. This potentially leads to greater and more persistent tissue damage, destruction of elastin, and the development and progression of emphysema.
When AAT-deficient individuals were reported to have increased susceptibility to emphysema development[2], researchers quickly recognised the balance between AAT and a destructive enzyme/s[3] (subsequently identified as a feature of neutrophil serine proteinases[9,26]) was key to maintaining elastin homeostasis and that emphysema reflected a disturbance in this balance where proteinase activity prevails.
The proteinase/antiproteinase imbalance between AAT and its cognate proteinase is reflected by the excessive amount and activity of neutrophil elastase in AAT-deficient individuals[27]. Neutrophil elastase load in the lung tissue is directly associated with the pathological severity of emphysema[28]. Furthermore, the systemic footprint of neutrophil elastase measured as a neutrophil elastase-specific fibrinogen cleavage product (AaVal360) is significantly higher in AAT-deficient individuals and correlates with the severity and progression in the early stages of AATD lung disease[29,30].
In the most prevalent form of AATD, it is the replacement of Glu to Lys at position 342 (hinge loop region) of the Z variant AAT protein that increases the likelihood of spontaneous polymerisation[6]. As a consequence, retention in the liver reduces secretion and hence the plasma and lung concentration, increasing susceptibility to tissue damage by neutrophil serine proteinases. In addition, polymerised AAT aggregates can be found in the lung tissue[31], associated with the accumulation and activation of neutrophils in the localised areas halting their migration into the airways, and hence causing more localised tissue damage. Furthermore, after degranulation, proteinases can become bound to neutrophil cell membranes (especially in AATD[32]) and remain active while being resistant to AAT inhibition, which further complicates the control of local proteinase activity, especially for AATD individuals.
The role of neutrophil elastase in the pathophysiology of emphysema is widely recognised. However, emerging evidence suggests that Proteinase 3 may play a greater role in driving emphysema in AATD. Proteinase 3 is stored at 3-4 times greater concentrations in the azurophilic granule than neutrophil elastase[33]. In addition to replicating pathological changes similar to emphysema in animals[26,34,35], mathematical modelling indicated that Proteinase 3 possessed a greater potential for injury than neutrophil elastase diffusing over a greater radius for longer before reaching an enzyme inhibitor equilibrium, especially in AATD[23]. Neutrophils from AATD subjects also expressed more Proteinase 3 on the cell membrane[32], which is replicated by treating healthy neutrophils with deficient Z as opposed to normal M AAT plasma, suggesting that AAT itself modulates cell membrane localisation.
An additional factor influencing the role of Proteinase 3 is that SLPI (the predominant antiproteinase in airway secretion) is a poor inhibitor of Proteinase 3 and suggests that Proteinase 3 likely has a more important role in tissue destruction, especially in AATD[22-24]. This is supported by the persistent Proteinase 3 activity in lung secretions of AATD patients compared to those with non-deficient chronic obstructive pulmonary disease (COPD) even when elastase activity is undetectable[32,36] and the greater plasma concentrations of the Proteinase 3 activity footprint than that of the neutrophil elastase footprint as measured by the specific fibrinogen cleavage products[37].
Neutrophils and emphysema
Neutrophil serine proteinases, particularly neutrophil elastase and Proteinase 3 are essential to neutrophil migration through the lung interstitium[24,38]. The movement of neutrophils is facilitated by the mobilisation of proteinases to the leading edge of the cell[39]. During this process, the concentration of the released proteinases in the pericellular space overwhelms their inhibitors, causing an area of obligate destruction closely surrounding the neutrophils until the proteinases diffuse away and an equilibrium is reached[24,32]. In AATD, the destructive effect is amplified as cell surface proteinases are less controlled, potentially causing a greater area of damage[40].
Accumulation of neutrophils in the lung is well documented in AATD. This neutrophilic accumulation is in response to locally released chemoattractants. Leukotriene B4 (LTB4) is generated and released by alveolar macrophages in response to excess neutrophil elastase activity[41]. Similarly, CXCL8 can be released from epithelial cells in response to an elastase challenge[42]. Both these potent chemoattractants have been detected at high levels in lung secretions of AATD[43]. Additionally, tissue degradation products generated by elastase from elastin can also add to the chemoattractant signal gradient[44], providing an amplification loop to the lung neutrophilic response, thus enhancing tissue damage.
Understanding the whole pathway involved in tissue damage in AATD (See Figure 1) provides a series of potential therapeutic interventions to restore a physiological balance and protect the lung.
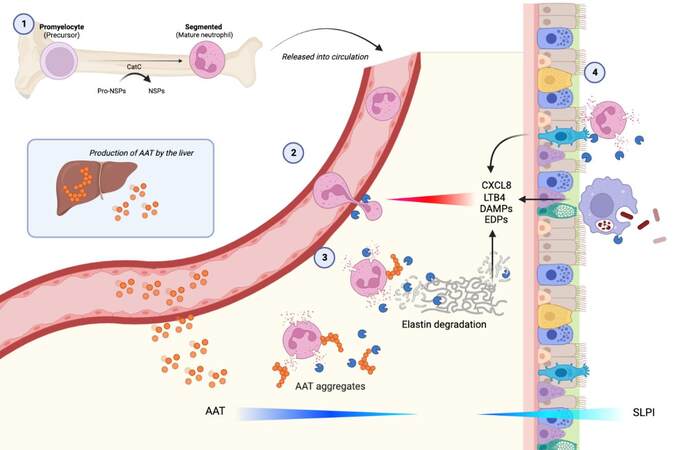
Figure 1. (1) Neutrophil proteinases are transcribed and packaged early in cellular differentiation in the bone marrow. Cell division later in differentiation divides the preformed packages (azurophil granules) amongst daughter cells and the mature cells are released into the circulation. (2) When the circulating cell senses a chemotactic gradient, it adheres to the endothelium via specific adhesion molecules and transmigrates through endothelial junctions. Granules migrate to the leading edge and the cell releases elastase at the point of transmigration, facilitating movement into the tissues, degrading a connective tissue pathway while leaving enzyme in its wake. (3) The presence of AAT polymers in AATD patients can potentially drive further neutrophil recruitment and local activation, which in the presence of deficiency amplifies local tissue damage, generating chemotactic fragments that add to the chemoattractant signal[31].
With this whole pathway in mind, there are several defined points at which the process can potentially be modulated.
Augmentation of the antiproteinase screen
Since the genetic deficiency is likely central to the disturbance of the physiological balance with neutrophil serine proteinase in the lung, the most logical step is to restore the missing protein to normal/safe levels. Gadek and colleagues[10] demonstrated this was feasible with AAT purified from plasma, given weekly at
Registry data found that patients receiving AAT augmentation had slower lung function decline, especially in a limited midrange detected by the forced expiratory capacity volume in 1 sec (FEV1) as a surrogate marker of emphysema progression[13,48]. This observation was supported by the slowing of lung density decline in later controlled clinical trials through measuring computed tomographic densitometry, a more direct marker for emphysema[15-17]. However, despite AAT infusion reducing the rate of lung density decline, a more recent randomised control trial did not observe an effect on halting deteriorating lung function stabilising or improving health status or reducing exacerbations[18], although the study was not statistically powered for such outcomes[49].
Assessment of the clinical efficacy of AAT augmentation therapy is complicated by different study designs and standard outcome measurements. The longstanding debate concerning the clinical impact of AAT augmentation therapy was more recently addressed by the European Respiratory Society. A comprehensive meta-analysis was undertaken and concluded a clinical benefit in reducing emphysema progression assessed by computed tomographic densitometry and the slowing of lung function decline[50], although a longer study of augmented and non-augmented patients showed no apparent benefit of augmentation as determined by the decline in FEV1[51]. Nevertheless, although the benefit of augmentation on the loss of lung tissue is now accepted, the progression continues even with this therapy, albeit at a slower rate, suggesting that other factors are at play[52].
Modified AAT forms are also being explored for possible improved stability and better cost-effectiveness. INBRX-101 (also known as rhAAT-Fc) is a modified AAT that is more resistant to oxidative inactivation[53]. Intraperitoneal administration of INBRX-101 in mice was more protective than using pooled plasma AAT in emphysema induced by elastases (pancreatic porcine elastase and neutrophil elastase) or cigarette smoke[54]. Recently, the phase 1 trial of intravenous INBRX-101 every three weeks showed that it restored functional AAT levels in the plasma of AATD patients, comparable to those of healthy individuals
ALTERNATIVE STRATEGIES FOR AATD INFUSION
Increase Alpha-1 antitrypsin secretion
Alpha-1 antitrypsin folding drug
In Z AATD, the reduced circulating levels of AAT are not due to a lack of production of AAT, but rather a result of intracellular Z AAT aggregation impeding secretion into the circulation. Based on this concept, drugs such as ZF874[56] and VX864[57] were developed to help rescue the misfolding of the Z AAT, thereby potentially preventing aggregation and increasing the release of monomeric AAT into the bloodstream and, thus, partially restoring inhibitory capacity. Phase 2 trials for both drugs have recently been completed. Results for both drugs are currently undocumented, although details relating to side effects have been released. It was announced that high-dose treatment of VX864 in patients with Z AATD over 28 days lowered plasma Z AAT polymers by approximately 90% and the level of functional plasma AAT was increased. However, the magnitude of this increase was deemed insufficient to protect the lung adequately[57]. A 48-week trial is currently underway to assess the long-term effects of VX864 in AATD patients.
Inhaled therapy
Delivering AAT directly to the airways might have a greater impact on airway inflammation than giving it intravenously, thereby focussing the distribution in the lung. The direct effect (if any) on emphysema remains unknown. A randomised controlled trial that assessed the effects of aerosolised AAT over 50 weeks in AATD patients with severe emphysema did not reduce the time to exacerbation (the primary outcome) compared to the placebo cohort[58], although post hoc analysis suggested a benefit on FEV1. While inhalation studies have shown that AAT levels rise in lavage fluid[59,60], the levels are unlikely to reach the alveolar region where emphysema occurs and subsequently penetrate through epithelial tight junctions into the interstitium.
Another strategy to control excessive proteinase activity would be to give SLPI, a proteinase inhibitor already enriched in airway secretions produced locally by bronchoepithelial cells[20]. SLPI also inactivates neutrophil elastase via direct 1:1 inhibition[61] and can additionally inhibit elastin-bound neutrophil elastase which is AAT-resistant[21,62]. SLPI levels in sputum or epithelial lining fluids are, however, influenced by local neutrophil elastase falling during inflammation[61,63]. This is also a feature of AATD as significantly lower levels of SLPI are present in the sputum of such individuals, likely a result of the increased local serine proteinase activity[36] and hence forming a potentiating inflammatory proteinase-rich loop.
SLPI has been given by inhalation for other conditions with excessive airway neutrophilic inflammation (such as in cystic fibrosis) showing an antiproteinase effect[64], but again in AATD where proteinase burden is high, a high concentration of SLPI will also have to reach the distal airways and penetrate into the interstitium. Furthermore, SLPI has no inhibitory activity on Proteinase 3[22] and Proteinase 3 also degrades SLPI[65], suggesting that SLPI may be less appropriate, especially as Proteinase 3 potentially has a major or even greater impact on driving disease progression than neutrophil elastase.
Neutrophil elastase inhibitors
Reagents (especially oral ones) capable of direct inhibition of neutrophil elastase would be a strong potential strategy based on the current understanding of elastase being a key direct mediator of emphysema and disease progression in AATD. Neutrophil elastase also orchestrates a series of proinflammatory responses, including cleavage activation of metalloproteinases[66], inducing the release of danger signals[67], and stimulating aberrant growth factor release[68] as well as impairing lung host defence mechanisms[69]. Thus, in addition to directly suppressing elastolysis, inhibiting neutrophil elastase could also prevent amplification of the inflammatory response and improve host defences.
AZD9668 is described as a potent selective inhibitor of neutrophil elastase[70]. Though the exact mechanism of action is currently unpublished, in vitro studies of AZD9668 successfully reduced plasma neutrophil elastase activity following whole blood stimulation by inhibiting both membrane-bound and liberated neutrophil elastase[70]. The disease-modifying potential of AZD9668 was validated in rodent models as oral administration of the drug attenuated systemic inflammation and neutrophil elastase-induced injury[70]. Furthermore, it was recently announced that oral administration of AZD9668 (Alvelestat or MPH966) successfully suppressed plasma evidence of neutrophil elastase activity in a phase 2 study in patients with Z AATD, demonstrating a progressive decline in the systemic fibrinogen biomarker of neutrophil elastase activity (AaVal360) in the high-dose treatment arm[71]. However, any effect on clinical outcomes has yet to be reported.
Modulation of neutrophils
Neutrophil chemotaxis
AAT-deficient neutrophils appeared to be inherently primed. When compared to healthy neutrophils, more AAT-deficient neutrophils spontaneously adhered to the endothelium[72] and displayed enhanced chemotactic response toward the chemoattractants LTB4 and CXCL8 which are abundant in the AATD lung[43]. The high chemoattractant burden, increasing neutrophil influx to the lung, and the resultant tissue damage suggest that modulating neutrophil migration to attenuate neutrophil-driven inflammatory effects could be advantageous. In the inflamed lung milieu, where a multiplicity of chemoattractants are present, chemokine receptor blockade offers an alternative approach to modulating elastase-mediated tissue damage.
CXCR2 is a membrane chemokine receptor expressed on neutrophils involved in regulating neutrophil chemotaxis. Preclinical studies investigating CXCR2 antagonism successfully prevented CXCL8-mediated chemotaxis[73]. The effect of CXCR2 blockade was also shown in a phase 2 trial with 615 COPD patients successfully reducing neutrophilic inflammation. However, more frequent episodes of exacerbation and pneumonia infections were reported in the high-dose treatment arms, implying that retaining adequate physiological responses is crucial for retaining host defence[74].
Neutrophil adhesion
An alternative approach to modulate neutrophil migration would be targeting endothelial adhesion. LTB4 mediates neutrophil adhesion by upregulating a b2-integrin (Mac-1) on endothelial cells as well as promoting neutrophil chemotaxis and degranulation[75,76]. Although LTB4 receptor antagonists have been studied in neutrophilic inflammatory diseases including cystic fibrosis and COPD, so far, no evident clinical benefit has been reported[77], even though Gompertz and Stockley[78] showed an anti-inflammatory effect. In addition, directly blocking the adhesion molecules could have the same effect, although both strategies run the risk of impairing this important secondary host defence as indicated above for CXCR2.
Proteinase production
A further approach would be to target upstream regulators required for maturating neutrophil serine proteinases before packaging into granules, thereby reducing the proteinase payload delivered per neutrophil. Cathepsin C (CatC) is an essential endopeptidase regulating the activation and subsequent packaging of neutrophil serine proteinases. An uncleaved signal peptide in the zymogens is signalled for degradation and when it is removed by CatC, the neutrophil serine proteinases become activated and packaged into the azurophilic granules. This packaging process occurs early in neutrophil maturation[79].
Recently, it was discovered that azurophilic degranulation is also significantly enhanced in AATD neutrophils through Rac2 signalling induced by elastase activity on the proteinase-activated receptor 2 on the neutrophils[80], adding further amplification to neutrophil elastase release. CatC is therefore an appealing target to simultaneously abrogate neutrophil serine proteinase activity and control the magnitude of degranulation. More importantly, CatC inhibition does not seem to interfere with neutrophil migration and antibacterial properties, as individuals with severe CatC deficiency (Papillion-Lefèvre syndrome) do not experience increased susceptibility toward major bacterial infections[81]. CatC blockade, therefore, should cumulatively reduce the quantity of neutrophil elastase and Proteinase 3 packaged into each azurophilic granule, lessen the amount of proteinase released and prevent excessive degranulation restoring a proteinase/antiproteinase balance to a level consistent with AATD while retaining neutrophilic defence properties. However, this should be balanced to avoid severe deficiency associated with the Papillion-Lefèvre syndrome.
CatC inhibition as a strategy is supported by AZD7986 (Brensocatib), a reversible CatC inhibitor used in a 24-week phase 2 trial in non-cystic fibrosis bronchiectasis where it reduced sputum neutrophil elastase activity by 40% while lowering exacerbation rates. In AATD, this approach may have not just been a beneficial effect on emphysema decline targeting both neutrophil elastase and Proteinase 3 but also an added benefit to the 30% of AATD patients with both emphysema and bronchiectasis[82].
SUMMARY
Neutrophil serine proteinases are responsible for driving inflammation and tissue destruction in the lung, especially in chronic lung diseases, such as AATD. This process is partly modulated by AAT replacement in AATD.
Replenishing AAT to restore protection against proteinase-driven damage is a logical and straightforward strategy but is costly and requires weekly infusions. Although AAT has broad immunomodulatory functions, it plays a passive role in preventing downstream injury as a side effect of excess neutrophil trafficking in response to chemoattractants. In comparison to the greater than $100,000 per patient per year for delivering augmentation therapy, novel oral drug candidates are likely to be cheaper but, importantly, less invasive and therefore more convenient for the patients.
CONCLUSION
Understanding the steps that lead to neutrophil migration, degranulation and tissue damage offers several potential strategies to modulate the proteinase/antiproteinase balance, although this has to ensure the important secondary host defensive functions are retained.
DECLARATIONS
Acknowledgments
Celine H. Chen would like to thank Dr Aaron Scott for his help with manuscript orientation.
Authors’ contributions
Both authors made substantial contributions to the conception and writing of the chapter.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Authors 2022.
REFERENCES
1. Sefton L, Kelsey G, Kearney P, Povey S, Wolfe J. A physical map of the human PI and AACT genes. Genomics 1990;7:382-8.
2. Laurell C and Eriksson S. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scandi J Clin Lab Invest 1963;15:132-140.
3. Eriksson S. Pulmonary emphysema and alpha1-antitrypsin deficiency. Acta Med Scand 1964;175:197-205.
4. Piitulainen E, Eriksson S. Decline in FEV1 related to smoking status in individuals with severe alpha1-antitrypsin deficiency (PiZZ). Eur Respir J 1999;13:247-51.
5. Lee JH and Brantly M. Molecular mechanisms of alpha 1-antitrypsin null alleles. Respir Med 2000;94 Suppl C:S7-11.
6. Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992;357:605-7.
7. Ogushi F, Fells GA, Hubbard RC, Straus SD, Crystal RG. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. J Clin Invest 1987;80:1366-74.
8. Lomas DA, Evans DL, Stone SR, Chang WS, Carrell RW. Effect of the Z mutation on the physical and inhibitory properties of alpha 1-antitrypsin. Biochemistry 1993;32:500-8.
9. Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. The induction of pulmonary emphysema with human leukocyte elastase. Am Rev Respir Dis 1977;116:469-75.
10. Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest 1981;68:1158-65.
11. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med 1987;316:1055-62.
12. Stockley RA, Dirksen A, Stolk J. Alpha-1 antitrypsin deficiency: the European experience. COPD 2013;10 Suppl 1:50-3.
13. Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Eur Respir J 1997;10:2260-3.
14. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. The Alpha-1-antitrypsin deficiency registry study group. Am J Respir Crit Care Med 1998;158:49-59.
15. Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999;160:1468-72.
16. Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009;33:1345-53.
17. Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of α-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res 2010;11:136.
18. Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. The Lancet 2015;386:360-8.
20. Abe T, Kobayashi N, Yoshimura K, et al. Expression of the secretory leukoprotease inhibitor gene in epithelial cells. J Clin Invest 1991;87:2207-15.
21. Morrison HM, Welgus HG, Stockley RA, Burnett D, Campbell EJ. Inhibition of human leukocyte elastase bound to elastin: relative ineffectiveness and two mechanisms of inhibitory activity. Am J Respir Cell Mol Biol 1990;2:263-9.
22. Korkmaz B, Attucci S, Jourdan ML, Juliano L, Gauthier F. Inhibition of neutrophil elastase by alpha1-protease inhibitor at the surface of human polymorphonuclear neutrophils. J Immunol 2005;175:3329-38.
23. Sinden NJ, Baker MJ, Smith DJ, Kreft JU, Dafforn TR, Stockley RA. α-1-antitrypsin variants and the proteinase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 2015;308:L179-90.
24. Campbell EJ, Campbell MA, Boukedes SS, Owen CA. Quantum proteolysis by neutrophils: implications for pulmonary emphysema in alpha 1-antitrypsin deficiency. J Clin Invest 1999;104:337-44.
25. Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)-antitrypsin deficiency (PiZ). Am J Respir Crit Care Med 1999;160:1968-75.
26. Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J Clin Invest 1988;82:1963-73.
27. Stockley R. Neutrophils and Protease/Antiprotease Imbalance. Am J Respir Crit Care Med 1999;160:S49-52.
28. Damiano VV, Tsang A, Kucich U, et al. Immunolocalization of elastase in human emphysematous lungs. J Clin Invest 1986;78:482-93.
29. Carter RI, Mumford RA, Treonze KM, et al. The fibrinogen cleavage product Aα-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011;66:686-91.
30. Carter RI, Ungurs MJ, Pillai A, Mumford RA, Stockley RA. The relationship of the fibrinogen cleavage biomarker Aα-Val360 with disease severity and activity in α1-antitrypsin deficiency. Chest 2015;148:382-8.
31. Mahadeva R, Atkinson C, Li Z, et al. Polymers of Z α1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol 2005;166:377-86.
32. Sinden NJ, Sapey E, Walton GM, and Stockley RA. S85 neutrophil cell membrane expression of proteinase 3 and its relationship to alpha-1-antitrypsin deficiency (A1ATD). Available from: https://thorax.bmj.com/content/67/Suppl_2/A41.3 [Last accessed on 9 Dec 2022].
33. Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol 2000;165:3366-74.
34. Borel F, Sun H, Zieger M, et al. Editing out five serpinA1 paralogs to create a mouse model of genetic emphysema. PNAS ;115:2788-93.
35. Gudmann NS, Manon-Jensen T, Sand JMB, et al. Lung tissue destruction by proteinase 3 and cathepsin G mediated elastin degradation is elevated in chronic obstructive pulmonary disease. Biochem Biophys Res Commun 2018;503:1284-90.
36. Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur Respir J 2013;41:1042-50.
37. Newby PR, Crossley D, Crisford H, et al. A specific proteinase 3 activity footprint in α(1)-antitrypsin deficiency. ERJ Open Res 2019;5:00095-2019.
38. Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 2003;163:2329-35.
39. Cepinskas G, Sandig M, Kvietys PR. PAF-induced elastase-dependent neutrophil transendothelial migration is associated with the mobilization of elastase to the neutrophil surface and localization to the migrating front. J Cell Sci 1999;112:1937-45.
40. Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 1995;131:775-89.
41. Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest 1991;88:891-7.
42. Walsh DE, Greene CM, Tomás PC, et al. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J Biol Chem 2001;276:35494-9.
43. Woolhouse IS, Bayley DL, Stockley RA. Sputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha-1 antitrypsin deficiency and the role of leukotriene B4 and interleukin 8. Thorax 2002;57:709-14.
44. Gaggar A, Weathington N. Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest 2016;126:3176-84.
45. Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun 2013;5:315-23.
46. McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the national heart, lung and blood institute registry of alpha 1-antitrypsin deficiency. alpha 1-antitrypsin deficiency registry study group. Chest 1997;111:394-403.
47. Liou TG and Campbell EJ. Quantum proteolysis resulting from release of single granules by human neutrophils: a novel, nonoxidative mechanism of extracellular proteolytic activity. J Immunol 1996;157:2624-31.
48. Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD 2009;6:177-84.
49. Stockley RA, Edgar RG, Starkey S, Turner AM. Health status decline in α-1 antitrypsin deficiency: a feasible outcome for disease modifying therapies? Respir Res 2018;19:137.
50. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J 2017;50:1700610.
51. Schouten IGM, Kasteleyn MJ, Tsonaka R, et al. Long-term effect of α(1)-antitrypsin augmentation therapy on the decline of FEV(1) in deficient patients: an analysis of the AIR database. ERJ Open Res 2021;7:00194-2021.
52. Stockley RA. Alpha-1 antitrypsin deficiency: have we got the right proteinase? Chronic Obstr Pulm Dis 2020;7:163-71.
53. Silberstein DZ, Karuppanan K, Aung HH, Chen CH, Cross CE, McDonald KA. An oxidation-resistant, recombinant alpha-1 antitrypsin produced in Nicotiana benthamiana. Free Radic Biol Med 2018;120:303-10.
54. Takeda K, Kim SH, Joetham A, Petrache I, Gelfand EW. Therapeutic benefits of recombinant alpha1-antitrypsin IgG1 Fc-fusion protein in experimental emphysema. Respir Res 2021;22:207.
55. Inhibrx Inc. Press Release. INBRX-101 shows favorable safety profile in patients with alpha-1 antitrypsin deficiency and demonstrates potential to achieve normal functional alpha-1 antitrypsin levels with monthly dosing. Available from: https://www.prnewswire.com/news-releases/inbrx-101-shows-favorable-safety-profile-in-patients-with-alpha-1-antitrypsin-deficiency-and-demonstrates-potential-to-achieve-normal-functional-alpha-1-antitrypsin-levels-with-monthly-dosing-301547416.html [Last accessed on 9 Dec 2022].
56. Centessa Press Release. Centessa pharmaceuticals reports financial results and business highlights for the second quarter of 2022; provides program update for ZF874. Available from: https://investors.centessa.com/news-releases/news-release-details/centessa-pharmaceuticals-reports-financial-results-and-business [Last accessed on 9 Dec 2022].
57. Vertex Press Release. Vertex announces primary endpoint achieved in phase 2 study of VX-864 in alpha-1 antitrypsin deficiency. Available from: https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-primary-endpoint-achieved-phase-2-study-vx-864 [Last accessed on 9 Dec 2022].
58. Stolk J, Tov N, Chapman KR, et al. Efficacy and safety of inhaled α1-antitrypsin in patients with severe α1-antitrypsin deficiency and frequent exacerbations of COPD. Eur Respir J 2019;54:1900673.
59. Hubbard RC, Brantly ML, Sellers SE, Mitchell ME, Crystal RG. Anti-neutrophil-elastase defenses of the lower respiratory tract in alpha 1-antitrypsin deficiency directly augmented with an aerosol of alpha 1-antitrypsin. Ann Intern Med 1989;111:206-12.
60. Kropp J, Wencker M, Hotze A, et al. Inhalation of 123I alpha1-protease inhibitor: toward a new therapeutic concept of alpha1-protease inhibitor deficiency? J Nucl Med 2001;42:744-51.
61. Sullivan AL, Dafforn T, Hiemstra PS, Stockley RA. Neutrophil elastase reduces secretion of secretory leukoproteinase inhibitor (SLPI) by lung epithelial cells: role of charge of the proteinase-inhibitor complex. Respir Res 2008;9:60.
62. Bruch M, Bieth JG. Influence of elastin on the inhibition of leucocyte elastase by alpha 1-proteinase inhibitor and bronchial inhibitor. Potent inhibition of elastin-bound elastase by bronchial inhibitor. Biochem J 1986;238:269-73.
63. Hill AT, Bayley D, Stockley RA. The interrelationship of sputum inflammatory markers in patients with chronic bronchitis. Am J Respir Crit Care Med 1999;160:893-8.
64. McElvaney NG, Nakamura H, Birrer P, et al. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest 1992;90:1296-301.
65. Rao NV, Marshall BC, Gray BH, Hoidal JR. Interaction of secretory leukocyte protease inhibitor with proteinase-3. Am J Respir Cell Mol Biol 1993;8:612-6.
67. Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 2016;16:378-91.
68. Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem 2003;278:2750-7.
69. Whitters D, Stockley R. Immunity and bacterial colonisation in bronchiectasis. Thorax 2012;67:1006-13.
70. Stevens T, Ekholm K, Gränse M, et al. AZD9668: pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J Pharmacol Exp Ther 2011;339:313-20.
71. Mereo BioPharma Press Release. Mereo biopharma announces positive top-line efficacy and safety data from “ASTRAEUS” phase 2 trial of alvelestat in alpha-1 antitrypsin deficiency- associated emphysema. Available from: https://www.mereobiopharma.com/news-and-events/news/2022/may/mereo-biopharma-announces-positive-top-line-efficacy-and-safety-data-from-astraeus-phase-2-trial-of-alvelestat-in-alpha-1-antitrypsin-deficiency-associated-emphysema/ [Last accessed on 9 Dec 2022].
72. Woolhouse IS, Bayley DL, Lalor P, Adams DH, Stockley RA. Endothelial interactions of neutrophils under flow in chronic obstructive pulmonary disease. Eur Respir J 2005;25:612-7.
73. White JR, Judithann ML, Peter RY, et al. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem ;1998.273:10095-8.
74. Lazaar AL, Miller BE, Donald AC, et al. for 205724 Investigators. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: a phase 2b trial. Respir Res 2020;21:149.
75. Tonnesen MG, Anderson DC, Springer TA, Knedler A, Avdi N, Henson PM. Adherence of neutrophils to cultured human microvascular endothelial cells. Stimulation by chemotactic peptides and lipid mediators and dependence upon the Mac-1, LFA-1, p150,95 glycoprotein family. J Clin Invest 1989;83:637-46.
76. Hoover RL, Karnovsky MJ, Austen KF, Corey EJ, Lewis RA. Leukotriene B4 action on endothelium mediates augmented neutrophil/endothelial adhesion. Proc Natl Acad Sci USA 1984;81:2191-3.
77. Bhatt L, Roinestad K, Van T, Springman EB. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin Immunol 2017;33:65-73.
78. Gompertz S, Stockley RA. A randomized, placebo-controlled trial of a leukotriene synthesis inhibitor in patients with COPD. Chest 2002;122:289-94.
79. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002;109:363-71.
80. Murphy MP, McEnery T, McQuillan K, et al. α(1) antitrypsin therapy modulates the neutrophil membrane proteome and secretome. Eur Respir J 2020;55:1901678.
81. Sørensen OE, Clemmensen SN, Dahl SL, et al. Papillon-lefèvre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest 2014;124:4539-48.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chen CH, Stockley RA. Targeting neutrophil serine proteinases in alpha-1 antitrypsin deficiency. Rare Dis Orphan Drugs J 2022;1:15. http://dx.doi.org/10.20517/rdodj.2022.18
AMA Style
Chen CH, Stockley RA. Targeting neutrophil serine proteinases in alpha-1 antitrypsin deficiency. Rare Disease and Orphan Drugs Journal. 2022; 1(4): 15. http://dx.doi.org/10.20517/rdodj.2022.18
Chicago/Turabian Style
Chen, Celine H., Robert A. Stockley. 2022. "Targeting neutrophil serine proteinases in alpha-1 antitrypsin deficiency" Rare Disease and Orphan Drugs Journal. 1, no.4: 15. http://dx.doi.org/10.20517/rdodj.2022.18
ACS Style
Chen, CH.; Stockley RA. Targeting neutrophil serine proteinases in alpha-1 antitrypsin deficiency. Rare. Dis. Orphan. Drugs. J. 2022, 1, 15. http://dx.doi.org/10.20517/rdodj.2022.18
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 2
likes
Like This Article 2
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.