
Burden of cerebral adrenoleukodystrophy on affected children and their families through the eyes of family caregivers
 0
0Abstract
Aim: This research assessed the needs of family caregivers of children with cerebral adrenoleukodystrophy (cALD), focusing on the diagnostic process; the burden of the disease on the child and their caregiver’s quality of life; and the physical, social, psychological, professional, and financial impacts on the whole family.
Methods: Family caregivers of children with cALD were recruited via the European Leukodystrophies Association International’s online platform, Leuconnect, to respond to a quantitative survey and a quality-of-life questionnaire and participate in a qualitative semi-structured interview. The questions focused on disease experience from onset to diagnosis and consequences on current life. Twelve family caregivers of 14 children were interviewed.
Results: cALD diagnosis took an average of 16.5 months, and 8 of 12 children were misdiagnosed, with parents often describing a lack of listening from doctors. Caregivers described bedridden children whose poor quality of life correlated with a high Neurologic Function Score. On average, they needed to care for their children 7.7 h/day, with serious consequences for their employment, social life, and psychological state.
Conclusion: Our interviews with family caregivers helped us to consider limiting diagnostic wandering by improving the skills of general practitioners and public knowledge of pathology. By gathering information on precise daily routines centered around a dependent child, we can better understand how to effectively support families by adapting not only the global care of the child but also to the following needs expressed for the entire family: better information, coordination of both care and administrative procedures, and real respite.
Keywords
INTRODUCTION
Cerebral adrenoleukodystrophy (cALD) is the most severe phenotype of ALD. This rare X-linked neurodegenerative disease results in the accumulation of very-long-chain fatty acids, which can lead to progressive and irreversible demyelination of the central and peripheral nervous system associated with adrenal insufficiency[1]. It is estimated that ALD affects approximately 1 in 20,000 to 1 in 15,000 boys[2,3].
Thirty-five percent of boys with ALD develop cALD, mostly between 2.5-10 years of age[1]. This evolution takes place in 3 phases, as described in Figure 1[4]: an initial, clinically asymptomatic latent phase, which progresses slowly; a second phase characterized by a significant and rapid progression of demyelinating lesions and by the first insidious appearance of cognitive deficits often leading to a vegetative state within 2 years after diagnosis[5]; and a stabilization phase during which the boys require full-time care. More than half of boys with cALD die within 5 years of diagnosis[6].

Figure 1. The three stages of cerebral adrenoleukodystrophy development.
If detected early enough, stabilizing the progression of the disease is possible with hematopoietic stem-cell transplantation (HSCT). This approach has shown a beneficial effect on clinical indices of disease and long-term survival, mainly in boys who are neurologically asymptomatic but who present with abnormal magnetic resonance imaging (MRI) findings and are treated very early[7,8]. At an advanced stage, the only possible treatments are corticosteroids to overcome adrenal insufficiency and palliative care, which is essential to improve the quality of life of the child and consists of combating pain, spasticity, and orthopedic complications associated with feeding by a gastric tube[4].
The physical consequences of cALD are well described[1,4,9], and different scales have been established to assess neurological damage on MRI (Loes score)[10] and clinical status [cALD Neurologic Function Score (NFS)][11]. The major functional disabilities (MFDs)[12] presented by children with cALD [Figure 1] have a direct impact on their quality of life. However, only two studies to date-Bessey et al. using the EQ-5D-5L questionnaire[13] and Beckman et al. using the pediatric PROMIS and Neuro-Qol questionnaires-have assessed the quality of life of children with cALD quantitatively[14]. A third study has focused on academic performance, absenteeism, and the need for academic assistance[15].
Regarding the burden on the family, two studies have assessed the impact of the disease on family caregivers in Taiwan[16] and Japan[17], respectively, using interviews and questionnaires completed by the parents of sick children. Questionnaires addressed difficulties encountered during diagnosis, the lack of curative treatment, problems linked to being a carrier of the disease, necessary and received assistance, and impacts on physical and mental health, social relations, and employment, and revealed both a negative impact of the disease in all of these aspects and an increased risk of depression for the parents.
No qualitative or quantitative data describing the disease burden of children with progressive cALD nor data comparing them to children who have received HSCT exist. Studies asking the caregivers about the overall consequences of their children's illness beyond its direct physical and medical implications are also lacking.
In this study, we assessed, through questionnaires and qualitative interviews, the impact of cALD on sick children and their families from the caregiver’s perspective. We described their experience with the disease since its onset and assessed both the burden of the disease and its treatment/care on the child and also the burden on the caregiver and the family in terms of physical, social, psychological, professional, and financial impacts. In addition, we assessed the quality of life of sick children and their caregivers and put into perspective the living conditions of children who received HSCT compared to those who did not. We discussed the difficulties encountered by these families and their children and the support that could be provided to them.
METHODS
Survey design
Data were collected in French (the mother tongue of the respondents) between December 2020 and January 2021 by telephone from family caregivers of sick children by two experienced investigators through the following means:
● A central questionnaire (average duration, 70 min) addressing living with the disease and the caregivers’ experience with medical, paramedical, and social care of their children, followed by standard quality-of-life questionnaires (EQ-5D-3L for the children and the RAND 36-Item Health Survey 1.0 for the caregivers), which were administered via the Leuconnect platform developed by the European Leukodystrophies Association (ELA) International. In addition to providing quantitative data, this central questionnaire highlighted the general context of living with the disease to refine the questions asked during the following qualitative interview.
● A qualitative interview lasting 40 min on average. A semi-structured discussion guide was used, containing open questions to collect the experience and deepen certain themes of the central questionnaire. The same themes were discussed as much as possible with all the respondents and included family experience with the disease, current situation, description of a typical day, screening and transmission, impact on projects, experience for the family, and avenues for improvement. However, the questions were systematically adapted to the respondents’ emotional state and personal history to spare their sensitivity.
The respondents were systematically informed of the presence of an investigator as a silent auditor who took notes and could be asked for clarification when necessary. The conversations were recorded, and the interviews were transcribed in full, with the respondent's agreement. Audio recordings were destroyed after the transcriptions were made.
The questionnaire and the discussion guide were designed from bibliographic research in collaboration with a multidisciplinary team familiar with the disease and in contact with families to ensure that the questions were consistent with the medical care of and the day-to-day experience with the disease.
Recruitment and screening of caregivers of cALD patients
The recruitment of caregivers and the collection of data were carried out online via the platform Leuconnect, which is dedicated to the management of an e-cohort of patients and their families concerned by leukodystrophies. The interface allows leukodystrophy patients and their relatives to register in view of personal health data collection and clinical study registration and participation. The interface also allowed investigators to enroll patients from the virtual cohort, accelerate recruitment, collect informed consent, and create online questionnaires. The sponsor of the study was responsible for the treatment of personal data collected during the study. The Leuconnect legal framework complies with the rules on the protection of individuals related to the treatment of personal data in compliance with the French modified law 78-17 of January 6, 1978 (“Loi Informatique et Liberté”), on computers, files, and liberties and the European General Regulation on Data Protection UE2016/619 RGPD. The ELA was responsible for processing the personal data collected using Leuconnect.
Caregivers of cALD patients applied spontaneously after information about the study was sent electronically to all volunteers (patients or caregivers) of the Leuconnect e-cohort.
All respondents were informed that their data would be collected completely anonymously, that they were participating freely in the study, and that they could withdraw their participation at any time. They electronically signed an information notice and informed consent form. It should be noted that, although the research adhered to strict ethical considerations of questioning people, an ethics committee was not consulted; this study in human and social sciences in the field of health did not fall within the scope of the definition of research involving humans as specified by article R1121-1 of the French public health code. The research was carried out under the French reference methodology MR-004 of the Commission Nationale Informatique et Libertés, guaranteeing the information and adequate processing of participants’ personal data.
The caregivers selected were adult volunteers residing in France who spoke fluent French and declared themselves to be or to have been the primary caregiver of 1 or more children diagnosed with cALD before the age of 18 years and less than 10 years ago. However, we decided to keep in the analysis the testimonial of a caregiver of 2 brothers who had both been diagnosed more than 10 years ago because it proved relevant to the study objectives to include data concerning the disease burden over a long period as a family caregiver.
A total of 12 caregivers caring for 14 children with cALD were interviewed.
Data analysis
A descriptive analysis of the quantitative data from the questionnaires was carried out. A consistency check was performed, and the recordings were replayed if there was any inconsistency in the responses. When not specified, quantitative data were represented by their mean and range of values. Children who had undergone HSCT were not differentiated from those who had not undergone HSCT in the section concerning the onset of symptoms and the diagnosis because their histories were similar. They were, however, separated for the sections concerning the children’s current life.
A thematic content analysis was carried out based on the qualitative data from the interviews: notes taken were completed with the analysis of the transcripts by the two interviewers and classified for each respondent according to the themes questioned, which were defined in advance by the study objectives and the knowledge of the disease and corresponded to the questions asked. However, if non-predefined themes were addressed by the respondents, a new category was created inductively. The content corresponding to each theme was then read “horizontally” across all interviews to draw general conclusions. The conclusions drawn from this analysis and the verbatim statements extracted from the interviews were used to illustrate and discuss the results from the quantitative analysis of the questionnaire.
EQ-5D-3L
Caregivers completed the standard EQ-5D-3L quality-of-life questionnaire for their children, which consisted of a descriptive section of 5 questions facilitating the evaluation of perceived problems concerning the 5 dimensions of mobility, self-care, current activities, pain and discomfort, and anxiety and depression; from here, a 5-digit health state profile (1 score per dimension) was converted into a utility value based on the preferences of the general French population[18]. A value of 1 point represented the best possible health and that of 0 points represented death, with negative values indicating worse than death. The score could not be calculated if a questionnaire response was missing. It was supplemented by a visual analog scale (VAS) on which caregivers were asked to rate their child’s current health, with 0 points indicating the worst possible health and 100 points indicating the best possible health. Scores were not submitted when the child was deceased.
RAND 36-Item Health Survey 1.0
Caregivers, including those whose children had died, also responded to the RAND 36-Item Health Survey 1.0 standard quality-of-life questionnaire assessing their quality of life in the last 4 weeks before the survey. This survey includes 36 questions divided into the following 8 dimensions: physical functioning, social functioning, physical pain, general health, energy/fatigue, role limitations due to emotional problems, role limitations due to physical health, and emotional well-being. Results were expressed in the form of scores from 0-100 points, with 100 points being synonymous with “good health”, calculated according to the method provided by the RAND Corporation[19].
RESULTS
Description of the population
A total of 12 caregivers caring for a total of 14 children with cALD were interviewed [Table 1]. All interviewees were the mothers of the children concerned, and 9 of 12 caregivers indicated that they were also carriers of the ABCD1 gene mutation. The average age of the caregivers was 44 years (range, 38-55 years). Children with cALD lived or had lived in the parental home in 13 of 14 cases. Two caregivers had 2 children with the disease. The respondents had been their children’s primary caregivers for an average of 4.2 years (range, 0.5-16.9 years).
Social-demographic characteristics of caregivers
| Total, n = 12 | |
| Sex | |
| Female | 12 |
| Male | 0 |
| Age | |
| 30-39 years | 2 |
| 40-49 years | 9 |
| 50-59 years | 1 |
| Relationship to the child | |
| Parent | 12 |
| Caregiver living daily with the child with cALD? | |
| Yes | 11 |
| No | 1 |
| Number of children in the family | |
| 1 | 2 |
| 2 | 5 |
| 3 | 5 |
| Number of sons with cALD | |
| 1 | 10 |
| 2 | 2 |
| Duration of care for a child with cALD | |
| < 1 year | 1 |
| 1-3 year (s) | 6 |
| 4-10 years | 4 |
| > 10 years | 1 |
| Level of education | |
| High school diploma | 3 |
| Vocational diploma, bachelor’s degree, or bachelor’s degree with honors | 8 |
| Master’s degree and above | 1 |
| Socio-professional category | |
| Craftsmen, traders, and business owners | 1 |
| Executives and higher intellectual occupations | 2 |
| Employees | 9 |
The mean age of the children at diagnosis and administration of the questionnaire was 8.3 years (range, 5.2-13.5 years) and 12.5 years (range, 8-28 years), respectively [Table 2]. Two children died before the survey (at the ages of 10 and 13 years). Two children had undergone HSCT. Of the 12 children who did not undergo HSCT, 10 children were completely dependent. Concerning the 2 caregivers with 2 children having the disease, 1 had a younger deceased son, and an older son, nearly 30 years old, who was still alive but completely dependent; the other had twin sons, 1 of whom was asymptomatic at diagnosis and underwent HSCT, while the other twin did not.
Characteristics of enrolled children with Cerebral adrenoleukodystrophy (cALD)
| Total, n = 14 | |
| Age at the time of the investigation* | |
| < 10 years | 1 |
| 10-12 years | 7 |
| 12-16 years | 5 |
| > 16 years | 1 |
| Age at diagnosis | |
| < 7 years | 2 |
| 7-9 years | 10 |
| > 10 years | 2 |
| Vital status | |
| Living | 12 |
| Deceased | 2 |
| Allograft received | |
| Yes | 2 |
| No | 12 |
First signs and diagnostic wandering
The caregivers told us that no child had been diagnosed with ALD before being diagnosed with cALD. The disease presented on average at the age of 6.7 years (median, 6.8 years; range, 3-12 years) with neurological signs among 11 of 14 boys [Table 3] and adrenal insufficiency for 1 boy (who presented with brain lesions on MRI scans performed at diagnosis). Two boys were diagnosed following the diagnosis of a brother, both in early stages of the disease but already showing brain lesions on MRI. Poor school performance and dysgraphia, or difficulty writing in general, were two disabilities that appeared in almost half of the children in whom neurological disorders were the first signs of the disease. In at least 7 of 11 cases, the caregiver mentioned the role of the school in reporting the disorders presented by the child. A caregiver indicated that the educational staff had requested a psychomotor assessment.
Types of disorders suggestive of cALD observed by parents in boys for which the first signs were neurological, n = 11
| Neurological signs | Total |
| Decline in school performance | 5 |
| Dysgraphia and other disorders associated with writing | 5 |
| Problems with vision | 3 |
| Behavioral disorders* | 3 |
| Hyperactivity | 2 |
| Reading disorders | 2 |
| Impaired motor skills | 2 |
| Spatial and/or temporal disorientation | 2 |
| Dyspraxia | 1 |
| Impaired motor coordination | 1 |
| Fatigability | 1 |
| Memory lapses | 1 |
| Trembling hands | 1 |
| Epileptic seizure | 1 |
| Vomiting and migraines | 1 |
The caregivers consulted a health care professional or visited the emergency room directly for their children no later than 4 months after the first signs of the disease appeared. In total, during this first consultation, almost half of the parents were told that there was no reason to worry and a third of the children were referred to a psychologist. Only 2 boys received a response allowing an immediate diagnosis [Table 4], i.e., MRI and referral to the emergency room. In one case, a drop in school performance and visual disturbances reported by the teacher resulted in the child being referred by the pediatrician to the pediatric emergency room. In the other case, a very good student became unable to read or write and was prescribed an MRI by the general practitioner.
Answers or referrals received by caregivers during the first consultation with a health care professional
| Answers | n | % (n = 12) |
| Nothing to report (“not to worry”) | 5 | 42 |
| Psychologist | 4 | 33 |
| Psychomotor monitoring | 2 | 17 |
| Speech therapist | 1 | 8 |
| Attending a center for language and learning disorders | 1 | 8 |
| Orthoptist | 1 | 8 |
| MRI | 1 | 8 |
| Pediatric emergency room | 1 | 8 |
According to caregivers, 8 of the 12 boys were initially misdiagnosed. For 3 of them, learning disabilities (including dysgraphia, dyslexia, and dyspraxia) were diagnosed. The other boys received diagnoses of encephalitis, hyperactivity, psychological trauma, a possible brain tumor, and gastroenteritis (for the boy for whom adrenal insufficiency was the warning sign). For at least 2 of the children, the disorders appeared at the time of a major family change (birth, death of a loved one). The regression observed in their development was associated with these events, sometimes for a long time, as in the case of 1 child for whom 2 years of wandering and the appearance of a motor disorder were necessary for him to be referred to a neurologist. Some parents also indicated that they thought their children’s troubles could be due to “school bullying”, “bad company”, or other “problems at school” and consulted a psychologist. The chronology of the children's diagnostic process is shown in Figure 2. It is important to note that the limits between the different stages of the disease were sometimes blurred for the caregiver in a process during which different signs of the disease may have appeared, sometimes in a very pronounced way or sometimes very progressively and without a specific date that the caregiver could remember. Moreover, the worrying signs were not necessarily new symptoms but could have been an amplification of earlier signs. On average, the age at diagnosis was 8.3 years (median: 8 years; range: 5.2-13.5 years), which was consistent with retrospective data collected from larger cohorts[7].
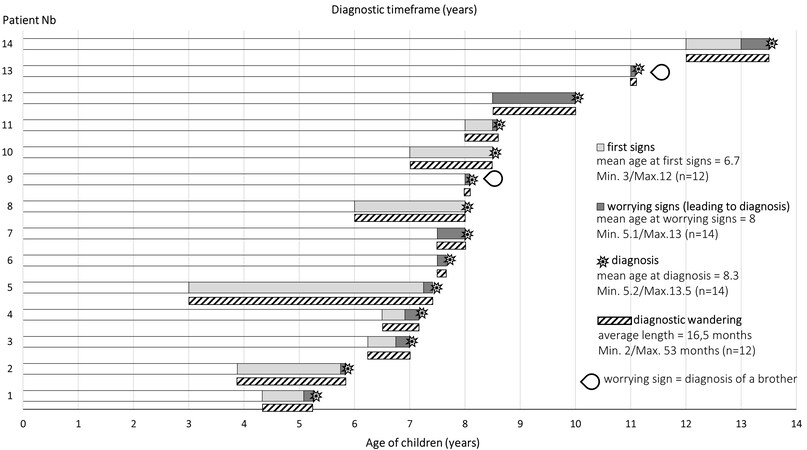
Figure 2. Chronology of the diagnostic process according to the parents of children with cerebral adrenoleukodystrophy. The age “at first signs” is the child’s age when, with hindsight, the caregiver saw the appearance of the very first signs of the disease. The age “at worrying signs” is the child’s age when the signs leading to a diagnosis appeared or when a brother was diagnosed in the absence of prior signs. n represents the number of children taken into account for each calculation.
During interviews, several respondents expressed their frustration at not being heard by health care workers when they started talking about their child’s difficulties, and the persistence required then to get an appointment with a specialist when they became concerned or felt they were not being taken seriously and/or accused of exaggerating the situation.
Interestingly, among respondents, 3 mentioned diagnoses of osteoarthritis or multiple sclerosis in the maternal grandmother of the sick children before the onset of cALD, 2 mentioned boys in the maternal family who died young from a brain issue or another unidentified reason, and a mother of a sick child had an adult brother with urinary disorders and unexplained loss of balance. Most of these diagnoses were subsequently attributed to ALD or Adrenomyeloneuropathy (AMN) in people still alive.
Diagnosis of the disease
When asked “How did you feel when your child was diagnosed with cerebral ALD?”, 1 in 2 caregivers reported a feeling of “incomprehension”. In addition, the words “disaster”, “catastrophe”, and “meltdown” used by respondents were indicative of the shock suffered. Some indicated that they would have preferred their children to have cancer because “everyone knows what it is” and because there would have been hope for a treatment. As in the Taiwanese study[16], a feeling of helplessness linked to the diagnosis was often expressed by caregivers. However, some reported a feeling of liberation for the parents and the children when they finally obtained a diagnosis but also, like the Taiwanese mothers, guilt at the idea of having reprimanded their children for their behavior when “it wasn’t his fault”.
Unlike Lee et al.’s study[11], in which mothers’ guilt over disease transmission arose immediately, in a particular context of beliefs and superstitions, guilt was expressed here only once by 1 mother
While the caregivers interviewed stressed the difficulty in finding the words to announce the diagnosis to their children, they did not spontaneously evoke a reaction of the children concerned or their siblings to the announcement of the diagnosis. When children with cALD were able to express a reaction, the feelings experienced, according to the caregiver, were primarily worry and/or sadness as they saw their parents sad and questioned about the disease progression as they were feeling diminished. Among siblings, according to the parent’s point of view, rather than reactions to the diagnosis itself, reactions to the disease progression were noted: they had lost their playmate, whom some siblings had sometimes decided to ignore. Four respondents mentioned sisters who did not want to talk about their brother’s illness, and 2 explained that their brother’s illness had oriented their daughters toward social or medical careers.
Burden on the child
Quality of life
Caregivers completed the standard EQ-5D-3L quality-of-life questionnaire for their children. The children’s difficulty levels in the five dimensions, as perceived by their caregiver, are shown in Table 5.
Responses to the questions in the descriptive part of the EQ-5D-3L questionnaire
| Mobility | Self-care | Current activities | Pain and discomfort | Anxiety and depression | |
| Children who did not undergo HSCT (n = 10) | |||||
| No difficulty | 1 | 1 | 2 | 3 | |
| Difficulties | 9 | 9 | 10 | 7 | 5 |
| No response | 1 | 2 | |||
| Children who underwent HSCT (n = 2) | |||||
| No difficulty | 2 | 2 | 2 | 2 | 1 |
| Difficulties | 1 | ||||
| No response | |||||
| All combined* (n = 12) | |||||
| No difficulty | 3 | 3 | 2 | 4 | 4 |
| Difficulties | 9 | 9 | 10 | 7 | 6 |
| No response | 1 | 2 | |||
According to their caregivers, the 2 children who underwent HSCT had no difficulty in terms of mobility, independence, or everyday activities and they felt neither pain nor discomfort. One of them was moderately anxious or depressed. In contrast, the 10 untreated children had difficulty in their daily activities: 8 of 10 boys were unable to walk and could no longer independently wash or dress themselves; 7 of 10 boys had moderate to extreme pain or discomfort; and 5 of 10 boys had moderate to extreme anxiety and depression.
Nine utility values were calculated (i.e., with the exclusion of the 2 deceased children and 3 incomplete questionnaires) [Table 6 and Figure 3], with the value of 1 point representing the best possible health and that of 0 points representing death (negative values indicating worse than death). These scores varied from
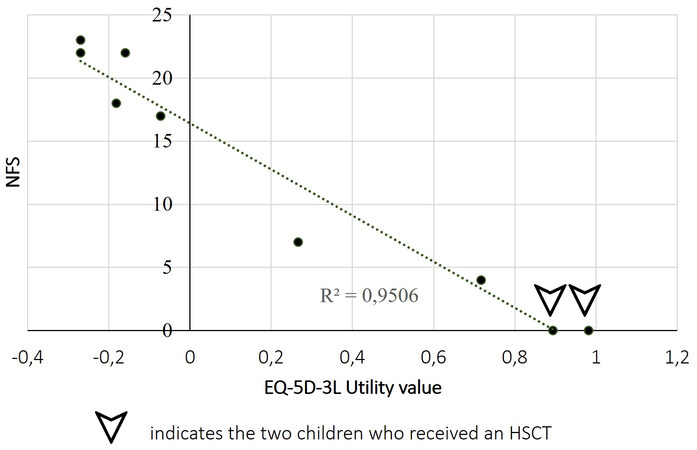
Figure 3. Correlation between the Neurologic Function Score and EQ-5D-3L utility value.
Comparison of children’s neurologic function score (NFS) and quality-of-life scores as assessed by the EQ-5D-3L questionnaire
| Years after diagnosis | NFS | Number of MFDs | VAS EQ-5D-3L | Utility EQ-5D-3L |
| 7.2 | 0* | 0 | 90 | 0.982 |
| 2.4 | 0* | 0 | 90 | 0.894 |
| 0.5 | 4 | 0 | 80 | 0.717 |
| 4 | 7 | 0 | 35 | 0.266 |
| 5.8 | 17 | 3 | 80 | -0.073 |
| 16.9 | 18 | 4 | 40 | -0.182 |
| 2.6 | 20 | 5 | Deceased | NC |
| 2.5 | 20 | 5 | 10 | NC |
| 2.5 | 21 | 5 | 40 | NC |
| 5 | 22 | 5 | Deceased | NC |
| 1.0 | 22 | 5 | 10 | -0.27 |
| 2.3 | 22 | 5 | 50 | -0.16 |
| 1.4 | 23 | 6 | 20 | -0.27 |
| 4.8 | 24 | 6 | 20 | NC |
The average health score assessed by caregivers on the VAS [Table 6] was 38.5/100 points for children who had not undergone HSCT versus 90/100 points for children who had undergone HSCT.
Symptoms of cALD and their impact on daily life
The symptoms observed by parents during the development of their children's disease included school difficulties according to 100% of the interviewed parents (e.g., decline in school performance, dysgraphia, speech disorders, problems with vision, and difficulty running), while 92% of the parents also mentioned spatial orientation problems, difficulty swallowing, and incontinence episodes [Supplementary Table 1]. The severity of the impairment of neurological functions was assessed by the cALD NFS[11] [Table 6], which is specific for cALD through 15 functional alterations and MFDs; notably, an NFS of 25 points represents a maximum loss of independence and a score of 0 points indicates the absence of obvious clinical signs.
None of the symptoms evaluated were observed in children who underwent HSCT. Ten children had 3 MFDs, including total incontinence, wheelchair dependence, and a complete loss of voluntary movement, to which loss of vision and the need to be fed by nasal tube or by gastrostomy were added for 6 of them. Figure 3 shows the direct correlation that exists between these functional alterations and the children’s quality of life as perceived by their caregivers.
The caregivers indicated that the 2 children who underwent HSCT and a recently diagnosed child were in mainstream schooling. Ten of 12 children who did not receive an HSCT were no longer in school, with the end of schooling occurring on average at the age of 9 years old. The median length of schooling after diagnosis was 8 months (range, 0-4 years). Four children lived in a specialized establishment, 3 of whom returned to the caregiver's home every evening. Seven children lived full-time in the caregiver’s home.
The parents spoke of a radical disease that develops with extreme rapidity. They described its development as being “overwhelming” and observed an “extremely rapid degradation” that took place in a few months or even a few weeks: “in three months, he... he lost everything”; “he is completely bedridden”; “today, there is nothing left”; and “he does not do anything consciously”.
Three parents indicated that their children did not express a reaction to this degradation, either because it had been too quick or because their cognitive impairment made the children appear completely careless. Concerning the children who were aware of their degradation and were able to express it, four children expressed, according to their parents, extreme sadness, a desire that “the disease would stop”, anxieties and questions about their loss of capacity, and then sometimes a sort of resignation to their fate and acceptance of the disability. One child refused at the onset of the illness to leave his home for fear that people would see him: “people will laugh at me”.
In contrast, the parents of the two transplanted children indicated that their children had a completely normal life and naturally used the term “cured”: “he knows that he is cured, he knows everything is fine”.
Medical and paramedical care
The 2 children who underwent HSCT had medical appointments 6 and 9 times per year, respectively, as part of the hematological and neurological follow-up. The non-transplanted children had an average of 4.5 medical or paramedical appointments per week (range, 2.3-8 appointments), including home visits and outpatient visits with physical and occupational therapy, speech therapy, and home care nurse aside from the home visits made by nurses as part of home hospitalization (5 children concerned). The medical personnel consulted and the frequency of consultations are presented in Supplementary Table 2.
Both children who underwent HSCT were followed up only by a hematologist 2 and 4 times a year, respectively, and 1 child by an endocrinologist 4 times a year, apart from the pediatric neurologist whom they also saw on average with the same frequency as children who had not undergone HSCT.
In the last year before the day of the survey, 7 children (including a child who underwent HSCT) had been hospitalized on average 2.3 times/year (range, 1-5 times/year). This represents a median cumulative duration of 11 days per year (range, 2-120 days).
One of the transplanted children was treated with corticosteroid therapy. The other transplanted child was not taking any treatment.
Non-transplanted children regularly received an average of 5 different treatments (range, 2-9 treatments; dosages and frequencies not requested from caregivers), including corticosteroids, anticonvulsants and/or antiepileptics, painkillers, and muscle relaxants for 12, 10, 9, and 6 children, respectively.
Burden on the caregiver
Quality of life for caregivers
Caregiver responses to the standard RAND 36-Item Health Survey 1.0 quality-of-life questionnaire are shown in Table 7. Caregivers generally perceived themselves to be in good general health (70/100 points) and were not particularly limited by their physical condition (88/100 points). However, they expressed fatigue (45/100 points), and some caregivers mentioned pain during interviews, especially back pain, which they attributed to having to carry their child. None of them mentioned a possible link to AMN, which can present as pain in female carriers of the disease who become symptomatic.
Scores of the eight dimensions of the RAND 36-item health survey 1.0 representing the current quality of life of caregivers, n = 12
| Dimension* | Average | Median | Min | Max | Standard deviation |
| Physical functioning | 88 | 91 | 73 | 91 | 5.6 |
| Role limitations due to physical health | 92 | 100 | 50 | 100 | 16.3 |
| Role limitations due to emotional problems | 67 | 100 | 0 | 100 | 45.0 |
| Energy/fatigue | 45 | 43 | 30 | 80 | 13.9 |
| Emotional well-being | 54 | 48 | 36 | 88 | 16.0 |
| Social functioning | 80 | 94 | 38 | 100 | 23.3 |
| Physical pain | 91 | 100 | 20 | 100 | 22.8 |
| General health | 70 | 73 | 30 | 85 | 15.6 |
The psychological impact of the disease was high among the caregivers interviewed (emotional well-being was evaluated at 54/100 points), in particular at the time of diagnosis, end of life, and death. Seventy-five percent of caregivers said they received or had benefited from psychological counseling for their sons’ disease. In more than half of the cases, the nursing staff initiated the counseling. One-third of caregivers took, at least occasionally, anti-depressant or anxiolytic treatments, and 5 of 12 declared that they had taken sick leave because of their psychological state (median number of days off, 30 days).
In everyday life with their children, the caregivers expressed incredible strength and a mindset that allowed them to cope psychologically and accompany their children on a daily basis. “I want things to go on, I want to move forward, I want to be well for my son, I want to be well for my husband and my daughter, and quite simply because I want to be well for me too”.
Impact on the daily life of caregivers
Apart from the 2 children who underwent HSCT and no longer required daily care, all of the caregivers interviewed expressed that they spent more time with their children at home than usual due to their state of health. The average time spent by caregivers specifically because of the illness was
Regarding their social life, 83% of caregivers declared that they had less or completely stopped their leisure activities, and 67% indicated that they had less or no free time at all and took vacations less than before or not at all. Regarding socializing with close family and friends, while no caregiver reported having stopped seeing their friends and family, at least one-third of caregivers had restricted their social relations. A caregiver said she was “in prison in [her] home”.
Eight caregivers said they missed some elements of their life before the diagnosis; particular emphasis was placed on the notions of freedom and sporting activities. The caregivers expressed a need to tale a break, but experienced difficulty in having the peace of mind to entrust their children and were afraid of being too far away.
Eight caregivers had reduced their professional activity and 4 had completely stopped working. Two spouses had also reduced their activity, and 1 spouse had adjusted his working hours. In total, 7 mothers and 1 father had completely stopped working, at least temporarily (range, 8 months to 10 years). This situation did not only concern parents during the period in which their sons were severely disabled; a caregiver whose child had undergone HSCT had also ceased all activity for 2 years, and a mother whose child died could not resume her activity until 1 year after the death of her son. Two in 3 caregivers felt that their professional situation had been affected by taking care of a child with cALD often because they had to stop all activities, and two-thirds of them also thought that this had affected or would affect their potential for a promotion or career progression. However, the role of employers has often been underlined as positive in maintaining employment, and work was perceived as life-saving for some caregivers, allowing them to maintain a social bond and not become too isolated: “I needed to keep working if I didn’t want to go nuts (laughs), and I absolutely didn’t want to be a nurse, but a mom”.
Beyond the social role played by work, stopping working or adjusting working hours generated a loss of income for the household. The disease also entailed expenses mainly due to adapting housing and vehicles with specialized purchased or rented equipment (e.g., wheelchair, splints, patient lift, shower trolley, etc.) and purchasing health products not covered by social security, such as diapers, mattress protections, and moisturizing creams. According to the parents, the French support system and the aid obtained from administrations and associations, even if they require complex and lengthy procedures, allowed for correct reimbursement of costs as well as compensation for both the child's disability and the role of the caregiver. No fees were charged for children who had undergone HSCT.
Family impact and support
Among the 11 caregivers living as part of a couple at the time of the survey, half said they had relationship problems with their spouse because of their children’s illness. In the interviews, the verbatims concerning the couple were very heterogeneous. We perceived tension, particularly due to daily fatigue or disputes over major decisions to be made (HSCT, gastrostomy, etc.) and, at the same time, the need to be united to face the diagnosis.
Some caregivers mentioned the negative impact of the diagnosis on family relationships in general (e.g., temporary estrangement, need for solitude, lack of time to care for non-sick children, difficulties in supporting the extended family when it was necessary to support the immediate family, etc.). The impact of their brother’s illness on the daily life of brothers and sisters who were not sick was not spontaneously approached by caregivers, and references to siblings were rare when the caregiver was asked to describe their daily life. However, 2 of them mentioned the need for the brothers and sisters to escape to places where there is no talk of disability or illness; 1 spoke of the jealousy of a sister toward the sick child who
Nevertheless, the 12 caregivers reported that they had an impression of benefiting from a solid support network in the management of their child’s illness. All had sought or received help at some point during the course of the disease. Eleven of the 12 caregivers indicated that they had been helped by members of their family, and 8 out of 12 spoke of the help provided by the ELA at different stages of the disease and, in particular, the major importance of being able to discuss with other families: “very quickly, we were... we were well supported. Right away, the family united, friends united, they all experienced it with us, they have all been there with us, the school too... of children... helped us a lot, supported us a lot. [...]. We were very well supported at the hospital, by the ELA, so yes, it was very... very supportive”.
Needs expressed
When asked to tell us how the experience of the disease or daily life with the disease could be improved or what advice they could give to other parents, caregivers again spoke of the need to take a break and obtain help in carrying out administrative procedures, in particular by centralizing them so as not to have to consult several actors. The notion of information often came up, specifically information for local doctors, who do not all know about the disease and how to manage it, and for parents via a referent local contact to learn about the disease in general. Better general knowledge of the disease among doctors would allow caregivers to establish a diagnosis more quickly, and more knowledge of the disease among the general public could improve the understanding of their life choices and their daily lives among friends and colleagues. So-called “comfort” care and alternative medicine were also mentioned, and a request for the centralization of needs in the event of a problem and the coordination of care was made.
Parents stressed the role of associations and the importance of being able to converse with other parents of children with cALD, whether to advise them on administrative procedures, offer them psychological support, or even establish a social link.
DISCUSSION
In this study, we described precisely, quantitatively, qualitatively, and individually the history of cALD from the point of view of the caregivers of sick children. We showed the value of interviewing the caregivers of sick children to enhance the knowledge of the disease and better understand their family life when living with the disease on a day-to-day basis and their real needs.
The period preceding the diagnosis confirmed the delay in diagnosing this disease, where the first signs, often insidious, were either minimized by the medical staff (while the entourage and the school were rightly worried) or mistakenly taken for another, more frequent problem, such as an attention disorder, learning disorder, or psychological issue. The response given following the first consultation (“nothing to report” in 40% of cases; a psychological problem in 30% of cases) was striking and testified to a lack of listening to the parents. The average duration of diagnostic wandering of 16.5 months-which is longer than the 6-12 months observed by the Taiwanese mothers in the study by Lee et al. but considerably improved from the 9.9 years observed to diagnose ALD in 1993[20]-is still too long to be able to intervene in time, representing a loss of a chance for many children with this disease[16]. An important element is the onset of symptoms in children who had no problems before. However, diagnosis remains very difficult, and the constant delay in diagnosis supports the implementation of neonatal screening.
The description given by the parents of the state of health of their children and their daily life highlighted the glaring difference between the “normal life” of transplanted children, considered as cured by their parents, and the rapid and total loss of independence of non-transplanted children according to the various indicators of overall quality of life.
The questions the parents were asked confirmed that the burden of care is consequent, and the care provided is extremely variable from one child to another and over time depending on the stage of development of the disease, sometimes leaving parents with the impression of a lack of coordination and advice in the follow-up of their children. Although extensive literature exists on transplantation, few articles address the care given to boys who cannot benefit from it (palliative care). In France, national guidelines for diagnosis, clinical workup, and treatment for ALD patients will be published shortly, which should allow better care of children throughout the country.
The disease has significant consequences for caregivers and their families, who are forced to organize a very precise schedule for each day. Caregivers were mostly forced to reduce their workload or give up their job, whereas only 9 of 21 mothers changed or lost their jobs in the Japanese study by Kuratsubo et al.[17]. Caregiving in France can facilitate administratively and financially the role of the family caregiver. Caregivers also reported limiting their sporting and/or cultural activities and radically changing their social life. The dimension of the quality-of-life questionnaire concerning life and relationships with others included scores corresponding to the question, “During the past 4 weeks, has your physical or mental condition interfered with your social activities, such as visits to friends, family, etc.?”. As the interviews took place between December 2020 and January 2021, the period of curfew and/or lockdown linked to the coronavirus disease 2019 epidemic in France was included in the last 4 weeks. Although caregivers were told to disregard the epidemic in their responses, we cannot exclude a bias due to resultant general limitations in social activities, all the more so among families with children diminished by the disease.
As in the study carried out in Japan[17], the psychological consequences of these changes in caregivers’ lives due to the disease were glaring, confirming the consequences of their children’s disease on their health and the need to encourage and help caregivers to take care of their own physical and mental health[21]. As the Taiwanese caregivers pointed out, support from associations and peer helpers is essential[16]. Also, although the disease has an impact on the couple’s relationship, it should be noted that, for the most part, the couple’s state of mind showed solidarity against the disease, with them reporting they “feel stronger” and vowing to
The impact on brothers and sisters is considerable because of the difficulties in establishing or maintaining communication with a disabled brother but also due to the space that the disease takes up in the family and the time that parents must sacrifice for the disease. Only the parents’ point of view on this subject was questioned here, yet studies on the feelings of healthy siblings of children with fatal illnesses show the significant psychological consequences of a child's illness on their siblings in good health[23]. Questioning them directly would improve the care of siblings by taking into account their own experience of their brother’s illness.
Only mothers of sick children responded to the survey; they are mainly the ones in the family who take care of the children and ensure their daily life. Often, they are expected to take on the role of the family caregiver[24]. Also, they are more involved in responding to this type of survey, as shown by the 2015 rare disease observatory, where 94% of parents of minor children who responded were women[25]. It would nevertheless be interesting to know the point of view of the fathers and to identify other needs, given that the concerns and feelings of fathers and mothers of children with rare diseases are different[26]. In an X-linked disease for which the consequences for mothers can be significant when they develop AMN, it would also be interesting to deep dive into mothers’ feelings about their own risk of developing the disease or the symptoms that they may already feel, as very few spontaneously brought up the subject of their own illness.
Although this study provides new and precise information concerning the family’s experience with cALD, the possible limits of the analysis should be considered. The number of respondents may seem limited, but cALD is a rare disease with, according to the epidemiology, just 6-7 boys per year in France newly affected. Still, this number remains significant and is similar to those of studies carried out in Taiwan[16] and Japan[17]. All of the respondents also had a connection with the ELA, which is probably a reflection of the role played by the ELA in informing families about the existence of this type of study. Families supported by this association may also be better equipped to respond. Testimonials from isolated families could show other results, especially in terms of the daily burden for parents and their needs. However, it seems difficult to recruit caregivers who are isolated and therefore less informed. In addition, this is a national study, with the particularities of French medico-social care that avoid the issues of care access and cost for patients, which could be a real difficulty in other countries. All of these results may therefore not be generalizable to all families of children with cALD throughout the world.
We appealed, by a retrospective interrogation, to the memory of the respondents, thus limiting the precision of the declarations on events that took place a few years previously. Some children have endured intense suffering before being relieved with pain medication or have experienced extremely agonizing periods when they lose their abilities very suddenly. The point of view of caregivers in the current situation can therefore be biased by the previous state of health of their children and is completely dependent on the parents’ interpretation of the attitudes of their multi-disabled children.
Nevertheless, for the first time in France, we can see, through the eyes of the caregivers of children with cALD, the daily lives of these children and their families. We described the life of healthy children who first develop very few specific signs of the disease and that of their parents who are powerless to voice their concerns, leading to a long period before diagnosis. It is clear that the implementation of newborn screening would be the best way to monitor ALD patients and detect cALD as early as possible, but as long as it is not implemented, this raises the issue of improving local medical services’ knowledge of the disease, its consequences, and its management in order to promote earlier diagnosis, which could improve care for patients and their families. We highlighted the extremely rapid and painful degradation attributed to the disease that renders children completely dependent when they have not received radical treatment, such as an HSCT, and the radical alteration of their lives and that of the whole family with notable consequences on all aspects of daily life and mental health. Finally, we showed what concrete needs these caregivers expressed and how better social and medical care for caregivers could improve the daily life of the entire family.
DECLARATIONS
AcknowledgmentsThe authors would like to sincerely thank the sick children and their families for their trust, courage, and precious time they so graciously offered to participate in this survey.
The research project was carried out on the Leuconnect platform in partnership with ELA International (ELA 2020-P003). We thank the teams of the ELA (Family Support Center and Research Center) who were mobilized to facilitate the realization of this study.
Authors’ contributionsDesigned the study, performed data-acquisition and caregiver interviews, and analyzed the results: Podevin M, Thomas G
Participated in the recruitment process: Saunier Vivar E, Rochereuil H, Thomas G
Provided the voice of the advocacy group in the design of the study and technical support for the use of the Leuconnect platform: Saunier Vivar E, Rochereuil H
Provided a critical review of the manuscript: Sevin C, Saunier Vivar E, Yazbeck E, Clément A, Bignami F
Drafted the manuscript and provided a critical review of the manuscript: Sevin C, Thomas G, Vivar E, Yazbeck E, Rochereuil H, Bignami F, Clément A, Podevin M
Availability of data and materialsTo preserve the anonymity of the respondents and their children, the data produced and analyzed during this survey are not available.
Financial support and sponsorshipThe entire study was funded by bluebird bio (France) SAS.
Conflicts of interestThomas G and Podevin M are current employees of Argo Santé. Clément A and Bignami F are former employees of bluebird bio (France) SAS. Saunier Vivar E and Rochereuil H are current employees of ELA international, GEIE. Sevin C served as a consultant for bluebird bio but declares that there are no current conflicts of interest with bluebird bio or Argo Santé. All authors declare that there are no conflicts of interest.
Ethical approval and consent to participatePatients electronically signed a consent to participate in the Leuconnect e-cohort and the survey.
Consent for publicationElectronic informed consent for publication was obtained from the participants.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis 2012;7:51.
2. Stone RT, van Haren K. Natural history of brain lesions in X-linked adrenoleukodystrophy: on-again, off-again. Neurology 2020;94:1058-9.
3. Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 2001;49:512-7.
5. Cappa M, Bizzarri C, Vollono C, Petroni A, Banni S. Adrenoleukodystrophy. Endocr Dev 2011;20:149-60.
6. Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol 2007;6:687-92.
7. Raymond GV, Aubourg P, Paker A, et al. Survival and functional outcomes in boys with cerebral adrenoleukodystrophy with and without hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2019;25:538-48.
8. Mallack EJ, Turk BR, Yan H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: meta-analysis and consensus guidelines. J Inherit Metab Dis 2021;44:728-39.
9. Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol 2007;3:140-51.
10. Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol 1994;15:1761-6.
11. Moser HW, Raymond GV, Lu SE, et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch Neurol 2005;62:1073-80.
12. Miller W. Stem cell-transplantation therapy for adrenoleukodystrophy: current perspectives. J Neurorestoratology 2017;5:5-19.
13. Bessey A, Chilcott JB, Leaviss J, Sutton A. Economic impact of screening for X-linked adrenoleukodystrophy within a newborn blood spot screening programme. Orphanet J Rare Dis 2018;13:179.
14. Beckmann NB, Miller WP, Dietrich MS, Orchard PJ. Quality of life among boys with adrenoleukodystrophy following hematopoietic stem cell transplant. Child Neuropsychol 2018;24:986-98.
15. van den Broek BTA, Page K, Paviglianiti A, et al. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv 2018;2:49-60.
16. Lee TY, Li CC, Liaw JJ. The lived experience of Taiwanese mothers of a child diagnosed with adrenoleukodystrophy. J Health Psychol 2014;19:195-206.
17. Kuratsubo I, Suzuki Y, Shimozawa N, Kondo N. Parents of childhood X-linked adrenoleukodystrophy: high risk for depression and neurosis. Brain Dev 2008;30:477-82.
18. Chevalier J, de Pouvourville G. Valuing EQ-5D using time trade-off in France. Eur J Health Econ 2013;14:57-66.
19. RAND Corporation. 36-item short form survey from the RAND medical outcomes study. Available from: https://www.rand.org/health-care/surveys_tools/mos/36-item-short-form.html [Last accessed 3 Jan 2020].
20. van Geel B, Assies J, Haverkort E, et al. Delay in diagnosis of X-linked adrenoleukodystrophy. Clin Neurol Neurosurg 1993;95:115-20.
21. Haren K, Bonkowsky JL, Bernard G, et al; GLIA Consortium. Consensus statement on preventive and symptomatic care of leukodystrophy patients. Mol Genet Metab 2015;114:516-26.
22. Namkung EH, Song J, Greenberg JS, Mailick MR, Floyd FJ. The relative risk of divorce in parents of children with developmental disabilities: impacts of lifelong parenting. Am J Intellect Dev Disabil 2015;120:514-26.
23. Tay J, Widger K, Stremler R. Self-reported experiences of siblings of children with life-threatening conditions: a scoping review. J Child Health Care 2022;26:517-30.
24. Migliorini L, De Piccoli N. Challenging gender perspective in the community to promote well-being and health. J Prev Interv Community 2020;48:121-31.
25. Maladies Diseases Info Services. L’Observatoire resultats et analyses Le parcours de santé et de vie. Available from https://www.maladiesraresinfo.org/s%E2%80%99informer-sur-les-maladies-rares/observatoire-des-maladies-rares.html [Last accessed 3 Jan 2020].
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sevin C, Thomas G, Vivar ES, Yazbeck E, Rochereuil H, Bignami F, Clément A, Podevin M. Burden of cerebral adrenoleukodystrophy on affected children and their families through the eyes of family caregivers. Rare Dis Orphan Drugs J 2022;1:17. http://dx.doi.org/10.20517/rdodj.2022.13
AMA Style
Sevin C, Thomas G, Vivar ES, Yazbeck E, Rochereuil H, Bignami F, Clément A, Podevin M. Burden of cerebral adrenoleukodystrophy on affected children and their families through the eyes of family caregivers. Rare Disease and Orphan Drugs Journal. 2022; 1(4): 17. http://dx.doi.org/10.20517/rdodj.2022.13
Chicago/Turabian Style
Sevin, Caroline, Gaëlle Thomas, Elise Saunier Vivar, Elise Yazbeck, Hélène Rochereuil, Fabrizia Bignami, Aurore Clément, Marieke Podevin. 2022. "Burden of cerebral adrenoleukodystrophy on affected children and their families through the eyes of family caregivers" Rare Disease and Orphan Drugs Journal. 1, no.4: 17. http://dx.doi.org/10.20517/rdodj.2022.13
ACS Style
Sevin, C.; Thomas G.; Vivar ES.; Yazbeck E.; Rochereuil H.; Bignami F.; Clément A.; Podevin M. Burden of cerebral adrenoleukodystrophy on affected children and their families through the eyes of family caregivers. Rare. Dis. Orphan. Drugs. J. 2022, 1, 17. http://dx.doi.org/10.20517/rdodj.2022.13
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 1
likes
Like This Article 1
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.