
Elastase-dependent congenital neutropenia
 0
0 , ...
, ... Abstract
Congenital neutropenia, which refers to an inherited deficiency in neutrophils, is a rare pathologic condition that affects approximately 0.0001-0.0009% of the general population. While congenital neutropenia can result from mutations in approximately 30 genes, its leading cause is gain-of-function mutations in the ELANE gene, which encodes the neutrophil granule serine protease, neutrophil elastase. This review focuses on established and novel concepts in the genetic, molecular and cellular mechanisms underlying neutrophil elastase-dependent neutropenia, and discusses possible new avenues for neutropenia research as well as potential novel treatment options that target pathogenic elastase variants.
Keywords
INTRODUCTION
Congenital neutropenia is a genetic disorder in which there is a lower-than-normal number of neutrophils. In cases of severe neutropenia, circulating neutrophil counts, which are normally around 1500-8500/μl blood in healthy human individuals, drop below 500/μl, and in very severe cases, they can be as low as
TYPES AND MAIN CAUSES OF NEUTROPENIA
Chronic neutropenia is a result of hereditary bone marrow-failure syndrome or an acquired deficit of neutrophils[6]. Whereas acquired neutropenia can occur secondary to autoimmune diseases, chemotherapy or drug reactions, congenital (hereditary) neutropenia is a genetically heterogeneous group of primary hematologic conditions that are mainly characterized by an anomalous neutrophil developmental program (granulopoiesis) leading to insufficient production of mature neutrophils in the bone marrow and, subsequently, low neutrophil counts in the bloodstream[3]. In rare cases, such as WHIM syndrome, mature neutrophils fail to exit the bone marrow, which also leads to peripheral neutropenia[7]. Mutations in more than 30 genes, such as ELANE, HAX1, G6PC3, SBDS, G6PT, CXCR4, TAZ, VPS45 or CXCR4 have been identified so far as causative factors in inherited neutropenia[3,8]. Neutrophil differentiation block observed in congenital neutropenia can be associated with changes in the bone marrow, such as bone marrow fibrosis; thus, it is not necessarily always an autonomous neutrophil cell phenomenon[9]. However, cell-intrinsic neutrophil maturation arrest, as a primary cause of neutropenia, has been reported for at least some neutropenia genes[3]. Despite the diverse underlying genetic defects that likely affect the function of various subcellular compartments in neutrophil precursors, such as ER or mitochondria, the proteins encoded by these genes may share (a) signaling pathway(s). Disruption of these pathways at different stages, by defective products of neutropenia-causative genes, would then have similar hematopoietic manifestations[3].
Mutations of the ELANE gene are the most common genetic aberration, accounting for about half of the cases of severe congenital neutropenia (SCN). Whereas a hallmark of untreated SCN is either chronic or severe quantitative and qualitative deficit in neutrophils[6], ELANE mutations are also the main cause of milder cyclic neutropenia (CyN), characterized by episodes of neutrophil scarcity recurring in a cyclic fashion, usually every 3 weeks. CyN-related ELANE mutations, which are either different from or overlap with those occurring in SCN, are found in > 80% of CyN patients[10]. The majority of SCN and CyN cases arise due to germline mutations in the ELANE gene and are of a dominant nature, typically affecting only one ELANE allele, and leaving the other ELANE allele intact[1]. In addition to heterozygous, autosomal dominant ELANE mutations, other models of inheritance, including single gene autosomal recessive, X-linked and sporadic patterns, have also been described in congenital neutropenia in association with different genes. For example, the HAX1 gene that causes SCN, also known as Kostmann disease, is an autosomal recessive genetic mutation[3].
SCN and CyN remain incurable diseases, although in the majority of patients, these conditions can be kept stable by regular, subcutaneous infusions of granulocyte colony-stimulating factor (G-CSF). Neutropenia can evolve into myeloid malignances, such as myelodysplastic syndrome or acute myeloid leukemia (MDS/AML). For effective therapy, patients with SCN typically require high doses of G-CSF. Those patients who receive the largest doses of G-CSF appear to be at the greatest risk of progressing to myeloid malignances[11,12]. In patients unresponsive to G-CSF or with an increased risk of MDS/AML, hematopoietic stem cell transplantation might be a potential therapeutic option[3].
ELANE MUTATIONS LEADING TO NEUTROPENIA
During the promyelocytic stage of granulopoiesis in the bone marrow, NE is synthesized as zymogen and post-translationally activated in the neutrophil precursors [Figure 1A]. The NE activation steps include N-terminal trimming by cathepsin C, which is required for NE protease activity. In addition, NE may be modified at C-terminus by an as yet unidentified protease[1]. The proteolytically-processed C-terminal is thought to serve as a binding site for adaptor protein complex 3 (AP3), implicated in the transport of NE to lysosome-like granules[1,13].
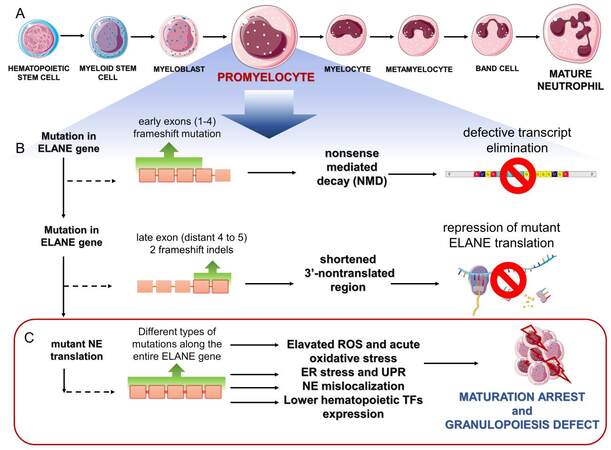
Figure 1. Outline of main steps leading to disruption of NE-mediated granulopoiesis. (A): Neutrophil development stages in the bone marrow, highlighting promyelocyte stage when differentiation blockade occurs. (B): ELANE frameshift mutations inducing cellular surveillance pathways that safeguard the quality of ELANE transcripts and prevent the production of mutant NE. (C): Mechanisms of mutant NE-induced neutropenia. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
NE mainly localizes to the primary granules of the neutrophil precursors, where it remains stored in a catalytically active, ready-to-use form. Neutrophils that are released from the bone marrow into the circulation, followed by recruitment to peripheral tissues, rely on catalytically active NE for their full spectrum of antimicrobial, pro-inflammatory and tissue-remodeling functions[14]. In addition to the localization of NE to the primary granules in the neutrophil precursors and mature neutrophils, the presence of this protease out of the granules, including in cytosol, at the cell surface and in the nucleus, has also been documented[15-17]. In mature, circulating neutrophils, transient release of NE from the granules is likely to be required for the physiological functions of these cells, such as generating and releasing neutrophil extracellular traps, or regulating neutrophil migration[15,17]. However, NE misplaced from the granules in the neutrophil precursors in the bone marrow is associated with neutrophil pathology, such as SCN (see below).
ELANE-based neutropenia results from the gain-of-function of mutant NE rather than diminished levels of native protein which might also be a consequence of monoallelic mutations[1]. This is supported by several lines of research. For example, a blockade of NE expression leads to the restoration of normal granulopoiesis and neutrophil differentiation, regardless of the type of underlying genetic mutation[18].
The ELANE gene consists of 5 exons and 6 introns, and > 200 mutations, distributed over all exons as well as some noncoding regions, have been described in association with congenital neutropenia[3,19]. However, to link genetic ELANE variants with pathologic outcomes, mechanistic studies are required. Mouse models of ELANE-targeted neutropenia have failed to accurately phenocopy human neutropenia because basal granulopoiesis was not disrupted in mice upon expression of abnormal murine NE, targeted at a position orthologous to human pathogenic NE mutation. A lack of suitable experimental neutropenia models has hampered the testing of ELANE mutants in vivo in the context of this pathology[20]. Human, murine or rat promyelocytic cell lines transduced with mutant ELANE, or patient neutrophil or patient-derived induced pluripotent stem cell (iPSC) lines, are typically used to unravel the biochemical and biological consequences of different ELANE mutations in myeloid progenitors. On the basis of these studies, several ELANE mutations have been identified to interfere with neutrophil maturation potential [Table 1].
Selected ELANE variants and their biologic consequences in experimental cell models
| Mutation variant | Protein change | Experimental model | Molecular and biological consequences [Ref] |
| NM_001972.4(ELANE):c.170C > T | A57V | SCN iPSCs | Diminished granulocytic differentiation[18] |
| NC_000019.10:g.852986A.T | I60F | SCN iPSCs | Elevated ELANE &NE expression, ROS levels & PML-NBs numbers; impaired neutrophil differentiation[26] |
| NM_001972.4(ELANE):c.211T > C | C71R | U937 cells | Protein processing changed, mistargeting, loss or reduction of proteolytic activity of NE[28] |
| HL60, NB4 cells; BM CD34 + cells | Increased ATF-6, ATF-4 mRNA & expression of ATF-6 targeted genes[25] | ||
| NC_000019.10:g.853345G.T | R103L | SCN iPSCs | Elevated ROS levels[26] |
| NM_001972.4(ELANE):c.353T > A (p.Ile118Asn)* | I118N | SCN iPSCs | Increased BIP and ATF-6 m RNA; decreased CEBPA and CEBPB mRNA, mislocalization of a proteolytically active NE, upregulated apoptosis, impaired granulocytic differentiation[30] |
| NM_001972.4(ELANE):c.416C > T | P139L | U937 cells | Loss or reduction in proteolytic activity of NE, increased ER stress and apoptosis[28] |
| SCN iPSCs | Elevated number of PML-NBs[26] | ||
| NM_001972.4(ELANE):c.452G > A | C151Y | SCN iPSCs | Diminished granulocytic differentiation[18] |
| SCN iPSCs | Increased ATF-6 mRNA, reduced granulocytic differentiation[38] | ||
| NM_001972.4(ELANE):c.640G > A | G214R (G185R) | murine 32D cells | increased GRP78 mRNA expression, unperturbed NE traffic, elevated apoptosis, diminished granulocytic differentiation[39] |
| SCN iPSCs | Increased ATF-4 mRNA, reduced granulocytic differentiation[38] | ||
| murine 32D cells, NB4 cells | unchanged ATF-4, ATF-6 mRNA level; reduced TF expression, mislocalization & loss or reduction in enzymatic activity of NE, viability not changed, impaired granulocytic differentiation[22] |
According to recent studies, a comprehensive gene editing screen in the human hematopoietic stem and progenitor cells (HSPCs) dissects the genetic bases of ELANE neutropenia pathogenicity. This model, in which HSPCs were edited with sgRNAs that targeted a broad range of ELANE mutants in vitro, faithfully recapitulated the known genetic features of ELANE-based neutropenia[21]. In addition, human-edited HSPCs have also been shown to produce an ELANE pathogenic variant-dependent abnormal hematopoietic engraftment function, following infusion into specific immunodeficient mice. Thus, these new xenograft models of gene-edited human HSPCs also enable the outcome of ELANE mutations to be tested in in vivo settings, which was not possible before with ELANE transgenic mice[21].
Most of these ELANE mutations are missense mutations, or in-frame insertion or deletions (indels), that lead to the expression of a mutant NE. Frameshift mutations that result in out-of-frame stop codons and nonsense mutations have also been described in association with ELANE neutropenia, but only involving distal exon 4 and exon 5 [Figure 1B]. These findings suggest that, when the premature stop codons are confined to the early ELANE exons, the NE protein is not translated[19]. A pooled CRISPR gene editing of the mutated early ELANE exons 1-4 and the mutated terminal ELANE exons (distal part of exon 4) and exon 5 in HSPCs have confirmed these predictions and revealed two different strategies that bypass neutrophil maturation arrest. The first mechanism was attributed to the elimination of erroneous mRNA that was transcribed from the mutated early exons (1-4), by a nonsense-mediated decay [Figure 1B]. This strategy prevented the translation of a mutant ELANE allele that contained premature stop codons in early exons, and was found to be a key determinant of ELANE variant pathogenicity. The other mechanism, which involved ELANE variants that escape a nonsense-mediated decay, resulted from a blockade of translation by a mutation (late exon -2 frame indels) that shortened the 3’-nontranslated region (3’-UTR) of ELANE[19]. Thus, a nonsense-mediated decay and repression of the mutated ELANE translation can rescue neutrophil precursors from maturation arrest [Figure 1B]. ELANE variants that bypass these rescue routes are translated, leading to deficiency in mature neutrophils. These findings are in line with clinical data that show only a -1, but not -2 frameshift mutation in terminal ELANE exons in patients with congenital neutropenia[19].
A knowledge of these two general escaping neutropenia rules suggests potential universal strategies for correcting pathogenic mutations, independent of ELANE neutropenia variants[8,19]. For example, to take advantage of nonsense-mediated decay, the corrective ELANE edits need to involve early exons. Another strategy may be based on placing corrective indels in late exons, and/or directly targeting the length and/or structure of the 3’-UTR to inhibit the translation of pathogenic ELANE variants.
MECHANISMS OF NE-MEDIATED NEUTROPENIA
The above findings help to explain the genetic bases of ELANE neutropenia. What remains less clear is how the presence of NE mutants following their translation from ELANE variants that avoid quality control mechanisms leads to a neutrophil shortage. Because NE is a protease, it is likely that neutropenia-causative ELANE mutations affect the enzymatic activity of NE. However, while most diminish or abrogate NE proteolytic potential, some mutations appear not to alter the proteolytic function of this protein[22,23]. Given that uncommon genetic alterations or biochemical features underlie the toxicity of NE mutants, at the molecular level, the pathogenic effects of NE variants might potentially be attributed to disruption of the NE protein structure, changes to NE subcellular distribution and/or altered interaction of mutant NE with other proteins, which overall affect the lifespan of neutrophil precursors, or their maturation or proliferation ability.
Several, not necessarily mutually exclusive, cellular mechanisms have been proposed to explain the impact of diverse NE mutations on promoting neutropenia. These include (i) NE misfolding followed by activation of the unfolded protein response (UPR) and apoptosis of neutrophil precursors; (ii) acute oxidative stress followed by upregulation of mutant NE levels; (iii) mutation-driven NE mislocalization; and/or (iv) a decreased expression of genes encoding key hematopoietic transcription factors as a result of the production of pathogenic NE variants [Figure 1C].
The most prevalent theory of how altered NE produces a neutrophil deficit is that mutations in ELANE result in improper folding, triggering the stress response pathway (UPR) within the ER, which leads to apoptosis of neutrophil precursors. The expression of incorrectly folded NE variants can be expected to culminate in promielocytes stage of granulopoiesis, when NE becomes the most abundant protein. The bulk of misfolded NE is, therefore, likely to accumulate at high levels in the ER and induce the UPR response followed by cell death. This mechanism is supported by experimental evidence, showing typical markers of the canonical UPR pathways, such as activation of molecular sensors (ATF6, CHOP) and master regulator of ER stress (GRP78), in cell models expressing pathogenic NE mutants[24,25].
However, not all neutropenia-causative NE variants are capable of consistently evoking the classic UPR response in experimental cell models. Degradation of misfolded proteins, independent of the canonical UPR, can also be a consequence of other protective cell strategies[26]. Recent findings point to elevated levels of promyelocyte leukemia protein nuclear bodies (PML-NBs), an indicator of acute oxidative stress, which may provide an alternative protective strategy in cells that harbor misfolding NE mutations. PML-NBs can be formed to diminish excessive ROS levels in the affected cells, but the presence of PML-NBs in cells with NE misfolding variants also stimulates cell metabolism and boosts the expression of NE, including NE mutants[26]. Thus, the formation of PML-NBs, independent of canonical UPR responses, may further promote neutropenic state.
Many NE mutations are predicted not to lead to NE misfolding, since they do not substantially interfere with the structure of the core protein[26]. Instead, they may be responsible for the failure of NE to be properly distributed within the cells. The first clue regarding how mutant NE can contribute to neutropenia and support for mislocalization theory came from studies in dogs[1]. Dogs suffer from CyN, which is similar to the human version, but canine CyN is autosomal recessive and does not result from NE but from AP3 mutation. AP3 is a protein involved in the trafficking of cargo proteins from the trans-Golgi network to lysosomes, suggesting that AP3 may play a role in NE trafficking and accumulation in granules. If NE, in order to be distributed in granules, interacts with AP3, a mutation in either gene could cause disruption to the intracellular trafficking of NE following the accumulation of NE in inappropriate subcellular compartments. An altered NE distribution in AP3-deficient dogs with canine cyclic neutropenia is in line with NE, which serves as an AP3 cargo protein[1]. Therefore, chain-terminating C-terminal NE mutations that disrupt NE interaction with AP3 could be expected to be mislocalized within the cells. This is what is observed in SCN patients[1].
Among other mistrafficking, NE mutations are, for example, NE variants that lack the ER localizing signal sequence, which is associated with transcriptional start site mutations[27]. The mistrafficking of NE and its accumulation in the cytoplasm beneath the plasma membrane or in cytosol has been reported for several NE variants[28-30]. Furthermore, cells that express the pathogenic G185R NE mutant associated with clinically severe forms of neutropenia, or that express shorter NE isoforms, exhibit a different distribution pattern of NE with localization to the nuclear and plasma membrane or nuclei[27,31]. Either proteolytically active or inactive NE mutants appear to be associated with off-site intracellular accumulation[22,28,30]. However, the proteolytic activity of these mislocalized NE variants has yet to be imaged in intact cells. The atypical intracellular distribution of NE might result in increased immunoreactivity for NE, a lack of control by intracellular inhibitors, and a lower activation threshold in neutrophils[16]. Taken together, these data suggest that the mislocalization of NE either via mutation of NE itself or in NE adaptive proteins, such as AP3 complex that regulates its transport into granules, can result in neutropenia.
Although in most cases, ELANE-based neutropenia is attributed to apoptotic death of promyelocytes, especially in the context of the UPR, mutant ELANE-dependent impaired differentiation of neutrophil precursors has also been proposed as a mechanism of neutrophil shortage. When human and murine myeloid cell lines with an inducible expression of human ELANE were used, suppressed expression of transcription factors that contribute to granulocytic differentiation was observed[22]. Since the suppression of these factors coincided with an altered subcellular localization of NE within the cells, these findings suggest that the mislocalization of the pathogenic NE variant may perturb the transcriptional regulation of granulopoiesis without affecting cell viability[22].
NE INHIBITORS IN THE DIAGNOSIS AND TREATMENT OF NEUTROPENIA
Despite the fact that neutropenia-associated NE mutations may or may not severely reduce NE proteolytic activity, blocking NE enzymatic activity using small synthetic NE inhibitors can correct NE localization to primary granules, which suggests that NE inhibitors are required for the proper localization of NE and points to the possible therapeutic or diagnostic potential of NE inhibitors[32]. Some specific low molecular weight inhibitors of NE have been found to positively affect the differentiation of ELANE-mutated cells. Inhibitors such as MK0339 or Sivelestat could improve the ability of the promyelocytic cells to differentiate[32]. Neutropenia patient-derived iPSCs are normally able to differentiate into granulocytes only in the presence of high doses of G-CSF, but when additionally supplemented with Sivelestat, granulocyte differentiation was possible with a much lower dose of G-CSF; this might be potentially beneficial for future therapies - lowering the chance of the development of myeloid malignancies. In addition, the presence of a small NE inhibitor restored normal intracellular NE localization[18,30]. NE inhibitor MK0339 was also able to correct the defective granulocytic differentiation of iPSCs and HL60 cells expressing mutant NE. Moreover, the inhibitor promoted the differentiation of both iPSC cells derived either from healthy donors or SCN patients[32].
Since NE and other serine proteases stored in neutrophil primary granules can contribute to tissue damage and severe pathology such as SCN, they have to be tightly controlled. However, despite the fact that neutrophils are a rich source of different protease inhibitors (PI), the role of these proteins in neutrophil development and/or function remains obscure. For example, neutrophils have long been shown to produce various PI, such as α-1 PI (SerpinA1), SLPI or elafin (PI3)[33,34]. Although proteolytically-active NE mutants, like unmutated NE, are vulnerable to inhibition with SerpinA1[23], the ability of other endogenous NE inhibitors to control the activity of NE mutants remains largely unknown.
More recently, many more PI genes have been found to be expressed in neutrophils, including SERPINB1, SERPINB2, SERPINB8, SERPINB9, SERPINB10, SERPINE1, SPINK1, WFDC5, and WFDC12[35,36]. The expression of some of these genes increases in long-lived neutrophils exposed to inflammatory stimuli, with elafin, SERPINB9 and SLPI most highly upregulated[35].
Even though the expression levels of the majority of these PI in neutrophil precursors remain to be determined, the deficiency of one of them, SLPI, has been found to be associated with SCN. SLPI expression is strongly reduced in myeloid cells from SCN patients carrying the ELANE or HAX1 mutations[37]. The treatment of myeloid cells with purified non-mutated NE highly increased the expression of SLPI. Likewise, silencing ELANE using specific shRNA has shown the dependence of SLPI expression on ELANE or HAX1 levels. These results suggest that NE is needed to induce SLPI expression in myeloid progenitors. The knockdown of SLPI in bone marrow progenitors inhibited the G-CSF-driven formation of neutrophils by decreasing the levels of genes responsible for proliferation and survival. These findings suggest that SLPI contributes to neutrophil production because it can regulate signal transduction downstream of G-CSFR in myeloid cells and improve their proliferation.
Another SLPI-dependent mechanism involved in neutropenia relates to the UPR response[25]. Correlative evidence links the severity of neutropenia, the expression of SLPI in myeloid cells from neutropenia patients, and the ability to trigger UPR stress pathways. These studies reported significantly lower levels of SLPI in patients with SCN compared to individuals with milder CyN[25]. Moreover, higher expression of SLPI in CyN has been suggested to provide protection against the UPR response. The combined suppression of SLPI and the transduction of ELANE mutant S126L in myeloid cells led to an elevation of the expression of several genes that are responsible for the induction of the UPR pathways. Taken together, these findings suggest that the magnitude of ELANE-triggered UPR response might be regulated by a natural inhibitor of NE - SLPI.
CONCLUSION
Gene editing technology has expanded our mechanistic understanding of how distinct ELANE mutations yield different outcomes, but has also potentially offered a universal approach for the treatment of ELANE neutropenia. In addition, previous experimental NE-based mouse models had limited clinical predictive power. The HSPCs models may offer a potential solution to this problem and provide a tool to further dissect the pathogenesis of congenital neutropenia. Finally, NE inhibitors need to be better explored in the context of neutropenia. A better understanding of the mechanisms underlying congenital neutropenia could help the development of alternative treatment options.
DECLARATIONS
AcknowledgementsNE structure shown in the graphical abstract was created using the PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC.
Authors’ contributionsMade substantial contributions to the conception and design of the study: Mazur A, Skrzeczynska-Moncznik J, Majewski P, Cichy J
Drafted the manuscript: Mazur A, Skrzeczynska-Moncznik J, Majewski P, Cichy J
Edited the manuscript: Cichy J
All authors approved the submitted version of the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis paper was supported by European Cooperation in Science and Technology (COST) Action CA18233, “European Network for Innovative Diagnosis and treatment of Chronic Neutropenias, EuNet INNOCHRON”.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Horwitz MS, Corey SJ, Grimes HL, Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am 2013;27:19-41, vii.
3. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primers 2017;3:17032.
4. Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis 2011;6:26.
5. Touw IP. Congenital neutropenia: disease models guiding new treatment strategies. Curr Opin Hematol 2022;29:27-33.
6. Papadaki HA, Mavroudi I, Almeida A, et al. Congenital and acquired chronic neutropenias: challenges, perspectives and implementation of the EuNet-INNOCHRON action. Hemasphere 2020;4:e406.
7. Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet 2003;34:70-4.
9. Vilboux T, Lev A, Malicdan MC, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N Engl J Med 2013;369:54-65.
10. Mir P, Klimiankou M, Findik B, et al. New insights into the pathomechanism of cyclic neutropenia. Ann N Y Acad Sci 2020;1466:83-92.
11. Dale DC, Bolyard AA, Shannon JA, et al. Outcomes for patients with severe chronic neutropenia treated with granulocyte colony-stimulating factor. Blood Adv 2022;6:3861-9.
12. Rosenberg PS, Zeidler C, Bolyard AA, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 2010;150:196-9.
13. Benson KF, Li FQ, Person RE, et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 2003;35:90-6.
14. Majewski P, Majchrzak-Gorecka M, Grygier B, Skrzeczynska-Moncznik J, Osiecka O, Cichy J. Inhibitors of serine proteases in regulating the production and function of neutrophil extracellular traps. Front Immunol 2016;7:261.
15. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010;191:677-91.
16. Skrzeczynska-Moncznik J, Zabieglo K, Osiecka O, et al. Differences in staining for neutrophil elastase and its controlling inhibitor SLPI reveal heterogeneity among neutrophils in psoriasis. J Invest Dermatol 2020;140:1371-1378.e3.
17. Zabieglo K, Majewski P, Majchrzak-Gorecka M, et al. The inhibitory effect of secretory leukocyte protease inhibitor (SLPI) on formation of neutrophil extracellular traps. J Leukoc Biol 2015;98:99-106.
18. Nasri M, Ritter M, Mir P, et al. CRISPR/Cas9-mediated ELANE knockout enables neutrophilic maturation of primary hematopoietic stem and progenitor cells and induced pluripotent stem cells of severe congenital neutropenia patients. Haematologica 2020;105:598-609.
20. Nanua S, Murakami M, Xia J, et al. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood 2011;117:3539-47.
21. Rao S, Yao Y, Soares de Brito J, et al. Dissecting ELANE neutropenia pathogenicity by human HSC gene editing. Cell Stem Cell 2021;28:833-845.e5.
22. Garg B, Mehta HM, Wang B, Kamel R, Horwitz MS, Corey SJ. Inducible expression of a disease-associated ELANE mutation impairs granulocytic differentiation, without eliciting an unfolded protein response. J Biol Chem 2020;295:7492-500.
23. Li FQ, Horwitz M. Characterization of mutant neutrophil elastase in severe congenital neutropenia. J Biol Chem 2001;276:14230-41.
24. Grenda DS, Murakami M, Ghatak J, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood 2007;110:4179-87.
25. Nustede R, Klimiankou M, Klimenkova O, et al. ELANE mutant-specific activation of different UPR pathways in congenital neutropenia. Br J Haematol 2016;172:219-27.
26. Olofsen PA, Bosch DA, Roovers O, et al. PML-controlled responses in severe congenital neutropenia with ELANE-misfolding mutations. Blood Adv 2021;5:775-86.
27. Tidwell T, Wechsler J, Nayak RC, et al. Neutropenia-associated ELANE mutations disrupting translation initiation produce novel neutrophil elastase isoforms. Blood 2014;123:562-9.
28. Köllner I, Sodeik B, Schreek S, et al. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 2006;108:493-500.
29. Liu Q, Zhang L, Shu Z, Ding Y, Tang XM, Zhao XD. Two paternal mosaicism of mutation in ELANE causing severe congenital neutropenia exhibit normal neutrophil morphology and ROS production. Clin Immunol 2019;203:53-8.
30. Nayak RC, Trump LR, Aronow BJ, et al. Pathogenesis of ELANE-mutant severe neutropenia revealed by induced pluripotent stem cells. J Clin Invest 2015;125:3103-16.
31. Massullo P, Druhan LJ, Bunnell BA, et al. Aberrant subcellular targeting of the G185R neutrophil elastase mutant associated with severe congenital neutropenia induces premature apoptosis of differentiating promyelocytes. Blood 2005;105:3397-404.
32. Makaryan V, Kelley ML, Fletcher B, Bolyard AA, Aprikyan AA, Dale DC. Elastase inhibitors as potential therapies for ELANE-associated neutropenia. J Leukoc Biol 2017;102:1143-51.
33. du Bois RM, Bernaudin JF, Paakko P, Hubbard R, Takahashi H, et al. Human neutrophils express the alpha 1-antitrypsin gene and produce alpha 1-antitrypsin. Blood 1991;77:2724-30.
34. Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol 1997;61:695-702.
35. Allaeys I, Ribeiro de Vargas F, Bourgoin SG, Poubelle PE. Human inflammatory neutrophils express genes encoding peptidase inhibitors: production of elafin mediated by NF-κB and CCAAT/enhancer-binding protein β. J Immunol 2021;206:1943-56.
36. Badola S, Spurling H, Robison K, et al. Correlation of serpin-protease expression by comparative analysis of real-time PCR profiling data. Genomics 2006;88:173-84.
37. Klimenkova O, Ellerbeck W, Klimiankou M, et al. A lack of secretory leukocyte protease inhibitor (SLPI) causes defects in granulocytic differentiation. Blood 2014;123:1239-49.
38. Dannenmann B, Zahabi A, Mir P, et al. Human iPSC-based model of severe congenital neutropenia reveals elevated UPR and DNA damage in CD34+ cells preceding leukemic transformation. Exp Hematol 2019;71:51-60.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mazur A, Skrzeczynska-Moncznik J, Majewski P, Cichy J. Elastase-dependent congenital neutropenia. Rare Dis Orphan Drugs J 2023;2:1. http://dx.doi.org/10.20517/rdodj.2022.12
AMA Style
Mazur A, Skrzeczynska-Moncznik J, Majewski P, Cichy J. Elastase-dependent congenital neutropenia. Rare Disease and Orphan Drugs Journal. 2023; 2(1): 1. http://dx.doi.org/10.20517/rdodj.2022.12
Chicago/Turabian Style
Mazur, Angelika, Joanna Skrzeczynska-Moncznik, Pawel Majewski, Joanna Cichy. 2023. "Elastase-dependent congenital neutropenia" Rare Disease and Orphan Drugs Journal. 2, no.1: 1. http://dx.doi.org/10.20517/rdodj.2022.12
ACS Style
Mazur, A.; Skrzeczynska-Moncznik J.; Majewski P.; Cichy J. Elastase-dependent congenital neutropenia. Rare. Dis. Orphan. Drugs. J. 2023, 2, 1. http://dx.doi.org/10.20517/rdodj.2022.12
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 0
likes
Like This Article 0
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.