
A report and review of the recurrent c.811C>T variant and mutation spectrum of Kindler syndrome in East Asians: a diagnostic odyssey of 2 weeks versus 49 years


Abstract
Kindler Syndrome (KS) is one of the rarest subtypes of epidermolysis bullosa (EB). It is characterised by congenital blistering, skin fragility, photosensitivity, and poikilodermatous skin changes. It is an autosomal recessive condition with an established disease-causing mechanism of having biallelic pathogenic variants in the FERMT1 gene. Multiple variants have been reported worldwide since the discovery in 1954. This case report describes two patients of Chinese descent with molecularly confirmed KS, one diagnosed in infancy while the other in mid-adulthood. It highlights the importance and clinical utility of diagnosing KS in children versus adults. The identification of recurrent c.811C>T variant in both patients also expedited the review of local databases and the existing mutation spectrum KS in East Asians.
Keywords
INTRODUCTION
Kindler syndrome (KS, MIM#173650) is a rare subtype of epidermolysis bullosa (EB) with autosomal recessive inheritance. About 400 cases have been reported worldwide to date since this syndrome was first described in 1954 by Theresa Kindler[1,2]. KS is characterized by skin fragility and blistering at birth, followed by the development of photosensitivity and progressive poikilodermatous skin changes. Other clinical characteristics include scarring in infancy, hyperkeratosis of palms and soles with fissuring, pseudosyndactyly associated with repeated blistering, and various mucosal manifestations such as ectropion, urethral stenosis, and labial leukokeratosis. Generalized poikiloderma eventually develops and persists throughout adult life in most affected individuals. KS’ phenotype is progressive and evolving during lifetime; skin blistering and photosensitivity occur less frequently with age[2]. Mucosal manifestations, including chronic gingivitis, dental caries, periodontitis, advanced periodontal bone loss and leukokeratosis of buccal mucosa, are frequent and prominent features in adulthood[2,3]. KS patients also have an increased lifetime risk of developing squamous cell carcinoma (SCC), with the cumulative risk of SCC increased to 67% by the age of 60, among which 54% will develop metastatic disease with high mortality[4].
The KS determinant gene, FERMT1, encodes a 677 amino acid protein, kindlin-1, a component of focal contacts in keratinocytes expressed in the epidermis, particularly in the basal keratinocytes[5,6]. It was reported that loss of kindlin-1 in keratinocytes leads to loss of cell polarity, decreased adhesion, reduced cell proliferation and increased apoptosis, resulting in abnormal skin fragility with defects in actin-extracellular matrix linkage[5,7]. It differs from classic keratin-extracellular matrix linkage underlying the pathology of other subtypes of epidermolysis bullosa[5]. It has also been shown that reduced production of kindlin-1 protein in keratinocytes is associated with altered mitochondrial structure, localization and function, making them prone to damage by oxidative stress, which could be a mechanism for susceptibility to the development of cancers[8].
KS is caused by the presence of biallelic pathogenic loss-of-function variants in FERMT1. Most reported FERMT1 variants associated with KS are null variants, while in-frame deletions and missense variants are proposed to be associated with milder clinical findings and later onset of complications[8]. Consanguineous marriage has been reported to be one of the major reasons for KS[9]. Individuals with a family history of KS or carriers of FERMT1 pathogenic variants are recommended to receive prenatal genetic testing, allowing early diagnosis in at-risk infants, which is pivotal for follow-up genetic counselling and early initiation of treatment, leading to improved long-term outcomes[2].
This case report summarises two Chinese individuals from Hong Kong who have the same pathogenic variant that has never been reported in East Asians. With the advancement in genomic sequencing technologies, while one patient received his molecular genetic diagnosis in infancy (patient 1), the other (patient 2) had suffered diagnostic odyssey for over 40 years. The present paper aims to outline the difficulties in differentiating KS from other dermatological conditions, and the significance and clinical utility of identifying the disease-causing genetic variants in children and adults with KS.
CASE REPORT
Patient 1
Patient 1 was an 11-month-old boy, born at full term via elective Caesarean section, with a normal birth weight of 2.875 kg. Antenatal history was unremarkable, including a normal routine second-trimester structural ultrasound. He is the second son of non-consanguineous Chinese parents, and his elder brother (2 years older) was reported to be healthy since birth. There is no contributory family history of skin and/or other related conditions.
The initial presenting features were two small round superficial skin desquamative lesions over the buttock, 2 cm and 1 cm in diameter, respectively, noted upon newborn examination immediately after birth. Subsequently, further extensive skin erosions developed all over the trunk and limbs, some spontaneously with no apparent trigger, but most significantly over pressure sites, which corresponded to the locations of blood pressure cuff application and blood sampling [Figure 1].
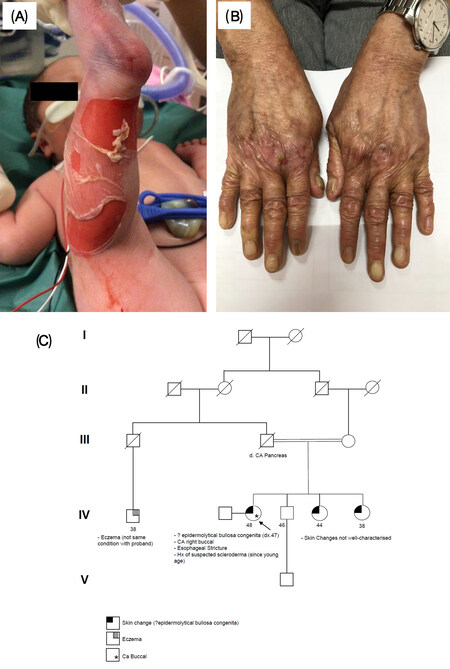
Figure 1. Clinical photos and pedigree of patients in this case report. A: patient 1 - extensive skin desquamation and erosions, particularly overpressure regions such as the site of blood pressure cuff; B: patient 2 - generalized skin xerosis with atrophic changes, dystrophic nails and patches of erosion; C: pedigree of patient 2.
In view of the extensive dermatological lesions with desquamation and exposure of the underlying raw dermal area, the baby was referred for clinical genetics evaluation and was transferred to the Neonatal Intensive Care Unit (NICU) of a tertiary hospital on the first day of life, where dermatological, paediatric surgical and plastic surgery burns unit support were available. Skin biopsy showed subepidermal blisters with a completely detached blister roof, basket weave keratosis and mostly intact slightly thickened epidermis. There were no features of inflammation, scarring, cytoid bodies, basal vacuolar degeneration, or caterpillar bodies. Special staining showed focal areas with linear hyaline basement membrane along the blister floor with no mucin deposition. Direct immunofluorescence was suboptimal due to completely denuded epidermis but no staining of immunoglobulins, complements or fibrinogen. The anatomical pathologist’s initial impression was a cell-poor subepidermal blistering disease, possible junctional type epidermolysis bullosa.
Trio-based rapid whole exome sequencing (WES) was performed by the corresponding author’s team in the Department of Paediatrics and Adolescent Medicine, The University of Hong Kong. The results returned positive with a turnaround time of 11 days. Compound heterozygous pathogenic variants in the FERMT1 gene were identified in patient 1. The first variant was FERMT1(NM_017671.4):c.811C>T p.(Arg271Ter), inherited from his father; and the second variant was FERMT1(NM_017671.4):c.1718+2T>C, inherited from his mother. Both variants had been previously reported in individuals with KS, and were classified as pathogenic according to The American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015)[10]. The genetic diagnosis of KS and supportive findings were communicated with dermatologists and anatomical pathologists, and both agreed that the clinical presentation and biopsy features were compatible.
The baby continued to receive in-patient multidisciplinary care. Emollients and dressing were applied for skin protection, and the dermatological condition gradually improved despite occasional scattered lesions and blistering, mostly corresponding to pressure or friction. The ophthalmological assessment confirmed no involvement of the eyes.
The early perinatal course was otherwise uneventful, except for transient respiratory distress after delivery which required antibiotic coverage and heated humidified high-flow oxygen nasal cannula (HHHFNC) for three days. However, soon after feeding started, the baby developed intermittent episodes of blood and mucus in stool with no increase in inflammatory markers and no infective pathogen isolated, which resolved when enteral feeding was withheld but recurred when feeding resumed. There was no consistent response to antibiotic treatment or switching to hypoallergenic formula. Recurrent sore buttock complicating frequent episodes of diarrhea was also documented. At 1 month old, there was an episode of severe hyponatraemia down to 110 mmol/L, presenting with repeated vomiting and decreased general condition but no seizures, which was corrected over 3 days. It was concluded to be likely secondary to poor feeding tolerance and significant gastrointestinal loss, after all, workup investigations returned unremarkable. There was also one episode of skin exudative infection due to methicillin-sensitive Staphylococcus aureus (MSSA), which was treated with a standard course of topical mupirocin. The baby was eventually discharged from hospital at 2.5 months old, with frequent loose stool but no longer contained mucus or blood. The body weight had dropped to below 3rd percentile, but there was steady fair weight gain in the period prior to discharge with oral feeding well established.
The baby continued to be followed up in a paediatric dermatological specialist centre. There was one episode of a blood-filled blister formation over the left thigh at 3 months old, which was aspirated, but otherwise, the skin condition was largely stable on generous emollients and cautious skin care. Blood investigations at 3 months old revealed iron deficiency, which had been subsequently normalized with oral iron and multivitamin supplements. Catch-up growth was satisfactory, and the child’s body weight had reached 3rd-10th percentile upon the latest follow-up at 11 months old. There were no desquamative lesions or blisters, and the skin was generally well except for a few patches of mild eczema.
Patient 2
Patient 2 was a 49-year-old woman, who was the eldest of 4 siblings born to consanguineous Chinese parents who were first-degree cousins [Figure 1]. Her two younger sisters were also reported to have some skin conditions but were not well characterized and with no definite diagnosis. Her parents and younger brother were reported to be healthy in general. No further family history of significance was reported.
She was reported to have thin skin with wrinkling, photosensitivity, and easy abrasion or blistering since birth. Skin biopsy had been performed on her and her younger sisters when they were young, but no specific diagnosis was reached. She also had right conductive hearing loss during her adolescence, and later hearing tests also confirmed high tone hearing loss on the left side. She started to have progressive dysphagia and choking as a teenager. She recalled having difficulties swallowing harder food, e.g., fried and crunchy food, since the age of 14-15, eventually could no longer tolerate a solid diet since around 30 years old, and has begun to rely on a blender to convert food into puree. The clinical assessment confirmed that she had a narrow oral aperture and scars were observed over her oropharynx. Oesophageogastroduodenoscopy (OGD) showed a circumferential stricture at the cricopharyngeus with scarring, and the endoscope was unable to pass through. A contrast swallow study showed decreased oesophageal peristalsis with impaired distensibility, suggesting oesophageal dysmotility. Rheumatological workup only showed elevated anti-nuclear antibody levels, with no specific diagnosis.
Eventually, she had an episode of emergency admission due to obstructive sensation at the cricoid region following ingestion of a Chinese cake, and after urgent contrast computed tomography of the neck ruled out any abscess, OGD was performed, and dilatation was done at the known stricture site at the cricopharyngeus. Biopsy only showed squamous mucosa with mild keratinization. Subsequently, she received regular follow-up and interval OGD with dilatation at the cricopharyngeal stricture.
At 45 years old, she developed further symptoms with oral ulcers, hair loss, and progressive weight loss of 5 kg over several months. Upon referral to a joint rheumatology-dermatology specialist clinic, she was noted to have generalized skin xerosis, atrophic changes over digits and extensor surfaces, dystrophic nails, loss of skin fissure over palmar region, and fragile skin with easy erosions over trauma-prone regions, in addition to microstomia [Figure 1]. She was suspected of having dystrophic epidermolysis bullosa, and was therefore referred to Clinical Genetics, and prescribed copious emollients for the management of her skin condition. She developed persistent buccal ulcers with leukoplakia simultaneously, and the biopsy showed moderately differentiated squamous cell carcinoma. Wide local excision with selective neck dissection and flap reconstruction was performed, which confirmed localized right buccal mucosa squamous cell carcinoma with no lymph node metastasis and clear excision margins. Residual trismus causing further feeding problems was noted during recovery in addition to her esophageal stricture.
After assessment in the Clinical Genetics Clinic, she was referred to and sponsored by the Hong Kong Genome Project (HKGP) with whole genome sequencing performed. Referral criteria and details of the HKGP were documented and published by Chu et al. (2022)[11]. Results showed homozygous FERMT1(NM_017671.4):c.811C>T p.(Arg271Ter), which was one of the same variants identified in patient 1, and was classified as pathogenic according to the ACMG guidelines[10]. Parental testing confirmed that her mother was a carrier of the variant, but testing was not available from the paternal side as her father had already passed away.
DISCUSSION
This case report summarised the phenotype and genotype of two Hong Kong Chinese patients with Kindler syndrome. The clinical presentations of the two patients were compatible with the phenotypes described in previous literature. However, prior to genetic investigation, the actual subtype of EB was not differentiable by the referring clinicians due to the mimicry clinical presentation of KS to other subtypes of EB, hindering many cases of diagnosis[12]. Genetic testing in the two families helped to reach a molecular diagnosis leading to better clinical management. Patient 1 received his diagnosis just 2 weeks after birth, which prompted early management by the multidisciplinary team. This informed the characteristics of progressive poikiloderma and photosensitivity in childhood, which is not a feature of other subtypes of EB. The timely diagnosis also enabled the strengthening of sun protection for patient 1 as he grew up, as well as the surveillance for skin cancer and mucosal tumour starting in adolescence. On the other hand, patient 2 had a longer diagnostic odyssey, with the molecular diagnosis confirmed at 49 years old. At the time of testing, the spectrum of KS manifestation including mucosal involvement and SCC had been observed. The identification of the underlying genetic disease-causing variant helped with extending cascade testing to the two younger sisters with skin issues that were less characterised. Limitations in the clinical comparison between these two patients included the significant difference in age, and their different pathogenic mutation profiles, although the genotype-phenotype correlation for Kindler syndrome is currently not well established. The ongoing Hong Kong Genome Project may help identify more patients with the same condition in this locality with continued longitudinal follow-up, which may allow better characterization of the influence of earlier genetic diagnosis on the clinical courses of this disease.
Both patients in the case report harbour the pathogenic variant of FERMT1(NM_017671.4):c.811C>T p.(Arg271Ter) (heterozygous in patient 1 and homozygous in patient 2). This is a well-characterised variant that has been reported in multiple literature and disease databases[13-17]. It was described as a common variant found in a tribe residing in a small, isolated village in rural Panama, in which 26 patients were found to harbour homozygous c.811C>T, and the high prevalence in the population is due to the high rate of consanguinity[16,17].
The early phenotypic profile of Patient 2, who was also homozygous for c.811C>T variant, is similar to that described in the Panaman cohort and consistent with the classical KS phenotype. However, as most of the Panaman patients described were either children or young adults at the time of reporting, the comparison of adult/later complications is limited. The detection of this variant has also been observed in other geographic regions/ethnicities, as it has been reported in three Caucasian patients, one Indian patient, one patient from Germany, and two patients from Oman[13-15,17].
Despite the prevalence and identification of the c.811C>T variant in multiple individuals with KS, this variant has never been observed in East Asians, in both the disease cohort and the unaffected population. This variant is found to be absent from East Asian populations in gnomAD version 2.1.1 and 3.1.2 with population sizes of 9,977 and 2,604, respectively; while 17 heterozygous alleles are observed in other populations (11 in non-Finnish European, four in African/African American, and two in South Asian). As it is uncommon to observe two patients with such a rare disease harbouring the same variant in a small geographical region of Hong Kong (with an estimated population of around 7.51 million in 2019), while a carrier of the variant has never been seen in a large population database, we have further investigated the allelic frequency in local cohorts. By reviewing data from 1,700 individuals from three local cohorts (in-house WGS data from the Hong Kong Genome Project, in-house WES data from the University of Hong Kong, and an open variant database of WES data)[18], the allelic frequency of c.811C>T remained as zero. It is possible that these two patients may have been linked by a common ancestor who carried the variant multiple generations ago. Additionally, this variant is also observed in multiple populations of different ancestry; it is also possible that this site has a higher chance of mutation occurrence.
By reviewing the reported KS patients in medical literature, we describe the mutation spectrum of KS in East Asians. A total of 15 variants were identified in the FERMT1 gene in Chinese and Japanese populations [Figure 2]. Similar findings of loss-of-function variants being the predominant mutation type spanning across the FERMT1 gene have been observed, and consanguinity contributed to the identification of homozygous variants in multiple reports. The majority of the reported mutations in FERMT1 affected splicing, followed by frameshift variants, nonsense variants, large exonic deletions, and missense variants. A recurring homozygous splice site deletion of c.1089+1del was reported in four Japanese patients, with the exon-8-skipped in-frame transcript as the major product, resulting in a functionally defective truncated kindlin-1 protein[17,19,20]. The same variant was also reported in compound heterozygous with another nonsense variant c.1761T>A in another Japanese patient[19]. Two other splice variants (c.1139+1G>A and c.1861-1G>A) have been reported in homozygous state in two Chinese patients[21,22]. Homozygous frameshift mutations, including c.220delC in exon 3, c.994_995delCA in exon 8, and c.1885_1901del in exon 15, were reported in three Chinese patients, all of which were predicted to cause a premature stop codon[23-25]. Ohashi et al. observed homozygous nonsense variant c.1761T>A in a Japanese patient and Li et al. reported two compound heterozygous nonsense mutations (c.193C>T, c.277C>T) in a Chinese patient[23,26]. Large deletion caused by Matrix Attachment Region elements mediated homologous recombination or Alu-mediated homologous recombination has been reported in two Chinese patients born to consanguineous parents, one of which is a 3017-bp deletion mutation spanning exons 7-9 (g.63601_66617del) and the other one is a 17-kb homozygous deletion spanning the introns 1-6, both are presumed to cause nonsense-mediated mRNA decay[27,28]. Apart from null variants, a missense homozygous variant c.1343T>A of uncertain significance has been reported in a Chinese patient with a clinical presentation of atrophic erythema and telangiectasia[29].
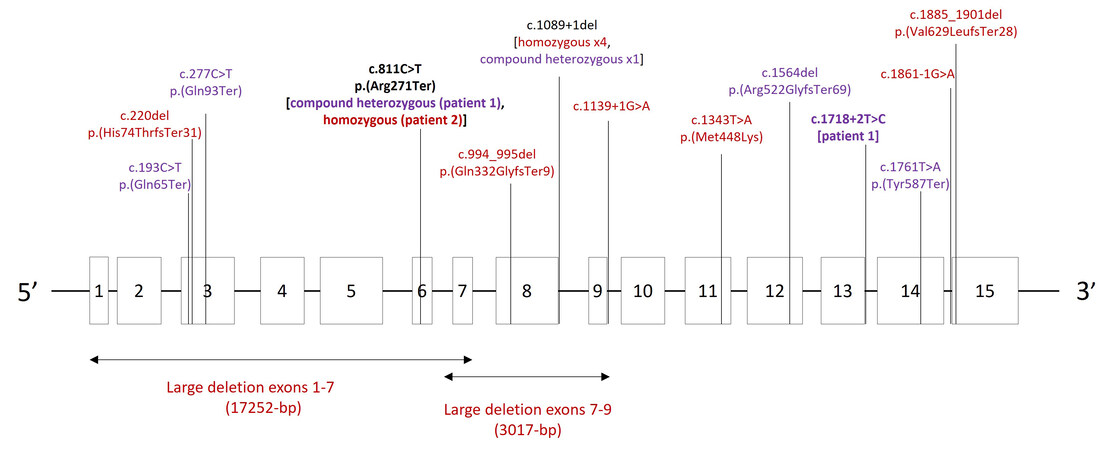
Figure 2. Mutation spectrum of East Asian patients with Kindler syndrome. Variants in bold were variants described in this case report in patient 1 and 2; variants in red were reported in homozygous states; variants in purple were reported in compound heterozygous with another variant.
We also expanded the investigation of the mutation spectrum of FERMT1 to patients of non-Chinese descent. A recent study by Li et al published in 2021 summarized 91 different pathogenic FERMT1 mutations in patients with Kindler syndrome[23]. After further review of Clinvar and LOVD databases, 27 additional pathogenic/likely pathogenic variants were identified in the FERMT1 gene, summing up to 118 variants. These 118 variants are scattered throughout the entire FERMT1 gene without obvious mutational hotspots, and most of which are loss of function variants, which is consistent with the existing understanding and the previous report by Li et al.[23]. Apart from the well-reported variant c.811C>T, there are several other notable FERMT1 pathogenic variants have been reported repeatedly. For example, c.676dupC p(Gln226fsTer16) is a recurrent variant in multiple populations with segregation data in six Pakistani families and one large Brazilian family[6,30-32]. c.910G>T p.(Glu304Ter) is another common variant observed in patients with different ethnic backgrounds, including Iranian, German, Australian, Caucasian, and Italian[6,32-35].
Since Kindler Syndrome is caused by the loss of kindlin-1 protein function, the variability of phenotype is not likely to be explained by the location of the mutation. A previous study that analysed clinical and genetic data of 62 patients also demonstrated that there is no genotype-phenotype correlation[14]. However, it mentioned that two patients carrying homozygous p.Arg100del or p.Ser400Pro showed a relatively mild phenotype and did not develop cancer, supporting the notion that incomplete expression or expressing mutated protein will cause milder phenotypes[14]. In the current study, patient 2 carries homozygous null variant c.811C>T p.(Arg271Ter), which is predicted to cause complete loss of protein activity, while patient 1 carries one null variant and one splicing variant c.1718+2T>C, possibly preserving certain truncated kindlin-1 expression. The difference in the type of mutation the two patients have might cause phenotype differences. However, a longer follow-up time for patient 1 and a larger scale functional study investigating the protein function of each specific variant are needed to systematically evaluate the genotype-phenotype correlation.
In conclusion, the present research team identified two patients (one in infancy and one at middle age upon the time of diagnosis) with molecularly confirmed KS of Chinese descent. Both patients carried the same pathogenic variant c.811C>T, which was otherwise not present in the controls and was unreported among KS patients in East Asian populations. This facilitated the review of local databases and the understanding of the existing KS mutation spectrum in East Asians, in comparison to other populations. This case report also succinctly discussed the clinical utility of genetic diagnosis in children versus adults, and contributed to the expedition of the effective clinical management of the two patients. Patients with an undiagnosed condition but high suspicion of underlying genetic etiology, in particular those with infantile onset of symptoms or strong family history of similar presentation, will benefit from early referral to clinical geneticists, and could possibly avoid prolonged diagnostic odyssey and allow early initiation of disease-altering treatment. Of course, for a more comprehensive comparison regarding phenotype and disease progression in patients with or without timely referral, we definitely need more future work utilizing a larger cohort covering patients diagnosed of different ages with longer clinical follow-ups. The adult patient found closure when the research team disclosed her WGS result and she said, “I could not believe you guys really did confirm an answer for me!”.
DECLARATIONS
AcknowledgmentsThe authors would like to express their greatest appreciation and gratitude to the patients and their family members for their selfless sharing and permission given to us to publish this important case report.
Authors’ contributionsConception and design: Chung BHY, Chu ATW
Drafting the article: Chu ATW, Chan JCK, Fung JLF
Data analysis and interpretation: Fung JLF, Tang W, Lee M
Critical revision: Chung BHY, Chu ATW
Final approval of the version to be published: Chung BHY, Chu ATW, Chan JCK, Fung JLF, Tang W, Lee M, Chung MH, Yu G, Li V, Ng CTH
Patient recruitment and data collection: Hong Kong Genome Project
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateEthical approval for this study was obtained from Central Institutional Board, Hospital Authority (HKGP-2021-001).
Consent for publicationWritten informed consent for publication from all patients was obtained.
Copyright© The Author(s) 2023.
REFERENCES
1. KINDLER T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br J Dermatol 1954;66:104-11.
2. Youssefian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews® [Internet]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK349072/ [Last accessed on 1 Mar 2023].
3. Handa N, Kachhawa D, Jain VK, Rao P, Das A. Kindler’s syndrome: a tale of two siblings. Indian J Dermatol 2016;61:468.
4. Guerrero-Aspizua S, Conti CJ, Escamez MJ, et al. Assessment of the risk and characterization of non-melanoma skin cancer in Kindler syndrome: study of a series of 91 patients. Orphanet J Rare Dis 2019;14:183.
6. Ashton GH, McLean WH, South AP, et al. Recurrent mutations in kindlin-1, a novel keratinocyte focal contact protein, in the autosomal recessive skin fragility and photosensitivity disorder, Kindler syndrome. J Invest Dermatol 2004;122:78-83.
7. Herz C, Aumailley M, Schulte C, Schlötzer-Schrehardt U, Bruckner-Tuderman L, Has C. Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J Biol Chem 2006;281:36082-90.
8. Ussar S, Moser M, Widmaier M, et al. Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet 2008;4:e1000289.
9. Youssefian L, Vahidnezhad H, Barzegar M, et al. The Kindler syndrome: a spectrum of
10. Richards S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
11. Chu ATW, Fung JLF, Tong AHY, et al. Hong Kong Genome Project. Potentials and challenges of launching the pilot phase of Hong Kong Genome Project. J Transl Genet Genom 2022;6:290-303.
12. Mariath LM, Santin JT, Schuler-Faccini L, Kiszewski AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol 2020;95:551-69.
13. Burch JM, Fassihi H, Jones CA, Mengshol SC, Fitzpatrick JE, McGrath JA. Kindler syndrome: a new mutation and new diagnostic possibilities. Arch Dermatol 2006;142:620-4.
14. Has C, Castiglia D, del Rio M, et al. Kindler syndrome: extension of
15. Kantheti P, Kubba A, Prabhu A, Batrani M, Hiremagalore R. Two novel mutations in
16. Penagos H, Jaen M, Sancho MT, et al. Kindler syndrome in native Americans from Panama: report of 26 cases. Arch Dermatol 2004;140:939-44.
17. Siegel DH, Ashton GH, Penagos HG, et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet 2003;73:174-87.
18. Ou M, Leung HC, Leung AW, et al. HKG: an open genetic variant database of 205 Hong Kong cantonese exomes. NAR Genom Bioinform 2022;4:lqac005.
19. Natsuga K, Nishie W, Shinkuma S, et al. Expression of exon-8-skipped kindlin-1 does not compensate for defects of Kindler syndrome. J Dermatol Sci 2011;61:38-44.
20. Wada M, Masuda K, Tsuruta D, et al. Case of Kindler syndrome resulting from mutation in the
21. Lin Z, Tan Y, Ma Z, et al. Ultrastructure of skin lesions and mutations in the FERMT1 gene in a patient with Kindler syndrome. Available from: http://www.pifukezazhi.com/CN/Y2010/V43/I10/677 [Last accessed on 23 Feb 2023].
22. Song D, Li Z, Liu J, Wang S. Kindler epidermolysis bullosa with pseudoainhum:a case report. Chin J Dermatovene 2022:36.
23. Li M, Li W, Zhu D, et al. Novel pathogenic mutations of FERMT1 in two Chinese Kindler syndrome families.
24. Meng L, Yang X, Wu Y, et al. A novel frameshift mutation in the
25. Oh SJ, Kim S-E, Lee SE, Kim S-C. Homozygous deletion mutation of the
26. Ohashi A, Kiniwa Y, Okuyama R, et al. A case of Kindler syndrome with severe esophageal stenosis. Int J Dermatol 2015;54:e106-8.
27. Gao Y, Bai JL, Liu XY, et al. A novel large deletion mutation of
28. Zhou C, Song S, Zhang J. A novel 3017-bp deletion mutation in the
29. Zheng BW, Zhu XZ, Lan Y, Ma JC, Li XQ. Unique variants in the
30. Martignago BC, Lai-Cheong JE, Liu L, McGrath JA, Cestari TF. Recurrent
31. Techanukul T, Sethuraman G, Zlotogorski A, et al. Novel and recurrent
32. Has C, Herz C, Zimina E, et al. Kindlin-1 is required for RhoGTPase-mediated lamellipodia formation in keratinocytes. Am J Pathol 2009;175:1442-52.
33. Kern JS, Herz C, Haan E, et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin-1 isoforms. J Pathol 2007;213:462-70.
34. Has C, Wessagowit V, Pascucci M, et al. Molecular basis of Kindler syndrome in Italy: novel and recurrent Alu/Alu recombination, splice site, nonsense, and frameshift mutations in the
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chu ATW, Chan JCK, Fung JLF, Tang W, Lee M, Chung MH, Yu G, Li V, Ng CTH, Hong Kong Genome Project, Chung BHY. A report and review of the recurrent c.811C>T variant and mutation spectrum of Kindler syndrome in East Asians: a diagnostic odyssey of 2 weeks versus 49 years. Rare Dis Orphan Drugs J 2023;2:2. http://dx.doi.org/10.20517/rdodj.2022.25
AMA Style
Chu ATW, Chan JCK, Fung JLF, Tang W, Lee M, Chung MH, Yu G, Li V, Ng CTH, Hong Kong Genome Project, Chung BHY. A report and review of the recurrent c.811C>T variant and mutation spectrum of Kindler syndrome in East Asians: a diagnostic odyssey of 2 weeks versus 49 years. Rare Disease and Orphan Drugs Journal. 2023; 2(1): 2. http://dx.doi.org/10.20517/rdodj.2022.25
Chicago/Turabian Style
Chu, Annie Tsz Wai, Joshua Chun Ki Chan, Jasmine Lee Fong Fung, Wenshu Tang, Mianne Lee, Man Ho Chung, Geoffrey Yu, Vivien Li, Calvin Tik Hei Ng, Hong Kong Genome Project, Brian Hon Yin Chung. 2023. "A report and review of the recurrent c.811C>T variant and mutation spectrum of Kindler syndrome in East Asians: a diagnostic odyssey of 2 weeks versus 49 years" Rare Disease and Orphan Drugs Journal. 2, no.1: 2. http://dx.doi.org/10.20517/rdodj.2022.25
ACS Style
Chu, ATW.; Chan JCK.; Fung JLF.; Tang W.; Lee M.; Chung MH.; Yu G.; Li V.; Ng CTH.; Hong Kong Genome Project.; Chung BHY. A report and review of the recurrent c.811C>T variant and mutation spectrum of Kindler syndrome in East Asians: a diagnostic odyssey of 2 weeks versus 49 years. Rare. Dis. Orphan. Drugs. J. 2023, 2, 2. http://dx.doi.org/10.20517/rdodj.2022.25
About This Article
Copyright

Data & Comments
Data


 Cite This Article 20 clicks
Cite This Article 20 clicks

 Like This Article 28
likes
Like This Article 28
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.