
Cathepsin K: both a likely biomarker and a new therapeutic target in lymphangioleiomyomatosis?
 0
0
Abstract
Lymphangioleiomyomatosis (LAM) is a rare disease that is characterized by cystic lung destruction and lymphangiomas and is associated with a high risk of osteoporosis-related bone fractures. Its diagnosis is based on pulmonary anatomopathological criteria combined with chest computed tomography. VEGF-D is the only serum diagnostic biomarker used in the clinic, while inhibition of the mTOR pathway by rapamycin is currently the only reference therapy for LAM. Human cathepsin K (CatK), a potent collagenase predominantly found in osteoclasts, is considered as a valuable target for anti-osteoporosis and bone cancer therapy. Recently, CatK, which is overexpressed in lung cysts, was proposed as a putative LAM biomarker. Moreover, CatK may take part in the LAM pathophysiology by participating in pulmonary cystic destruction and bone degradation. Accordingly, targeting collagenolytic activity of CatK by exosite-binding inhibitors in combination with mTOR inhibition could represent an innovative therapeutic option for reducing lung destruction in LAM.
Keywords
CATHEPSIN K: AN OVERVIEW
Introduction
Cysteine cathepsins (cathepsins B, C, F, H, K, L, O, S, V, X, and W) are a group of eleven structurally related papain-like proteases (clan CA, family C1) in humans (MEROPS database; http://merops.sanger.ac.uk). These enzymes are primarily found in acidic endosomal/lysosomal compartments where they partake in various cellular processes (i.e. non-specific intracellular proteins degradation and turnover, autophagy, and immune responses)[1,2]. Cysteine cathepsins, which are expressed either ubiquitously or with tissue and cell-type specificity, are also found outside lysosomes under specific conditions, contributing most often when they are dysregulated to a wide range of pathophysiological events[3,4]. Cathepsin K (CatK), which was discovered in the 1990s, is a highly potent collagenase with a restricted cell distribution. Its predominant expression in osteoclasts has promptly suggested specialized functions, including bone and articular cartilage resorption[5]. Moreover, CatK, which is overexpressed during osteoporosis as well in bone cancers, was validated as an attractive target for anti-osteoporosis therapy and anti-tumor treatments[6-10]. Meanwhile, CatK is gaining a growing interest according to potential other roles in physiological and pathological processes, including rare diseases.
First, we will summarize contemporary knowledge on molecular aspects of CatK (genomic organization, tissue expression, functional and structural characteristics, substrate specificity, regulation of its activity) and review interventional strategies currently developed to selectively prevent the uncontrolled collagenolytic activity of CatK in osteoporosis and bone cancer. Then, following a concise description of lymphangioleiomyomatosis (LAM), a rare human disease, we will discuss both the interest in using CatK as a new LAM biomarker and in targeting CatK for reducing lung destruction in LAM. Indeed, according to the severity of the disease and the ensuing loss of lung function that might be associated with CatK expression level in LAM, inhibition of CatK could be an additional therapeutic option, besides inhibition of the mTOR (mammalian target of rapamycin) pathway.
Cathepsin K: molecular and biological characteristics
Human CatK is encoded by a single gene (CTSK, ~12.1 kb), localized on chromosome 1q21, like cathepsin S (CatS)[1]. CTSK gene expression is organized and tightly controlled at multiple steps, and likely involves the interaction of several transcription factors that are activated by cytokines. For instance, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukins (IL)-6 and -13 positively regulate the synthesis of CatK, while transforming growth factor (TGF)-β1 and IL-10 may repress its expression level (for review:[5,11]).
CatK (EC 3.4.22.38) is a monomeric endopeptidase (~24 kDa), which shares the common papain-like structure, which consists of two globular domains folded together to give a ‘‘V’’-shaped active site cleft configuration in the middle. Albeit the catalytic triad Cys25, His159, Asn175 (papain numbering) is well conserved, mapping studies of the crystal structure of CatK revealed differences in the substrate binding subsites (labeled Sn-Sn’) that are located on both sides of the substrate’s scissile bond (corresponding to the complementary positions Pn-Pn’)[12]. Notably, residues forming the S2 binding pocket of CatK, its major determinant of substrate specificity, are crucial for its prevalence for hydrophobic and aliphatic amino acids (for review:[5]). Both binding pattern and substrate specificity of CatK were previously detailed by Lecaille et al.[5]. Importantly, CatK displays an unusual ability among cysteine cathepsins to accept Pro in the S2 pocket, a recognition residue of collagens. Based on these data, selective substrates and activity-based probes of CatK were developed[13-16].
Compared to other human proteases, CatK displays the unique potency to cleave within the triple helix of type I and II collagens[17]. Furthermore, the presence of chondroitin 4-sulfate (C4-S), a glycosaminoglycan (GAG) predominantly expressed in bone and cartilage, is required to promote its collagenolytic activity by forming high molecular complexes[18]. The formation of an active complex between the negatively charged C4-S and specific positively charged residues of CatK (and shaping a so-called exosite, which is located opposite the active site) is unique among human cysteine cathepsins [Figure 1]. In the absence of GAG, the ability of CatK to cleave triple helical collagen fibrils is impaired. Accordingly, it has been hypothesized that selective inhibitors able to disrupt CatK/C4-S complex by targeting the C4-S binding exosite could inhibit its collagenase activity without impairing other regulatory peptidase and protease activities (see further section). In addition to collagens, CatK hydrolyzes other extracellular matrix (ECM) components such as elastin fibers, and aggrecans[19]. CatK is a lysosomal protease that is mainly active at acidic pH 5.5 and rapidly inactivated at neutral pH. Nevertheless, lung macrophages and osteoclasts secrete CatK, which remains active in the pericellular space due to the presence of H+-ATPase pump or Na+/H+ exchanger[20,21]. In addition to its primary location in bones, high levels of CatK were also detected in synovial fibroblasts, aortic smooth muscle cells, macrophages, and epithelial cells, while CatK mRNA levels were increased in other tissues such as ovary, synovia, heart, skin, and lungs, following scarring or inflammation[10,22,23]. In addition to its collagenolytic activity and its involvement in ECM remodeling, CatK takes part in the maturation of thyroid hormones, participating in the processing of thyroglobulin[24,25]. Moreover, CatK plays a pivotal protective role in lung homeostasis via its ability to proteolytically inactivate TGF-β1[26,27]. In the last decade, a paradigm shift from the concept of “simple” protein-degrading enzymes to key signaling scissors was proposed[28], as demonstrated by the paramount importance of cysteine cathepsins (including CatK) in diverse cell signaling cascades (e.g., PPAR-γ/caspase-8-mediated cell apoptosis, TGF-β signaling pathway)[29-33].
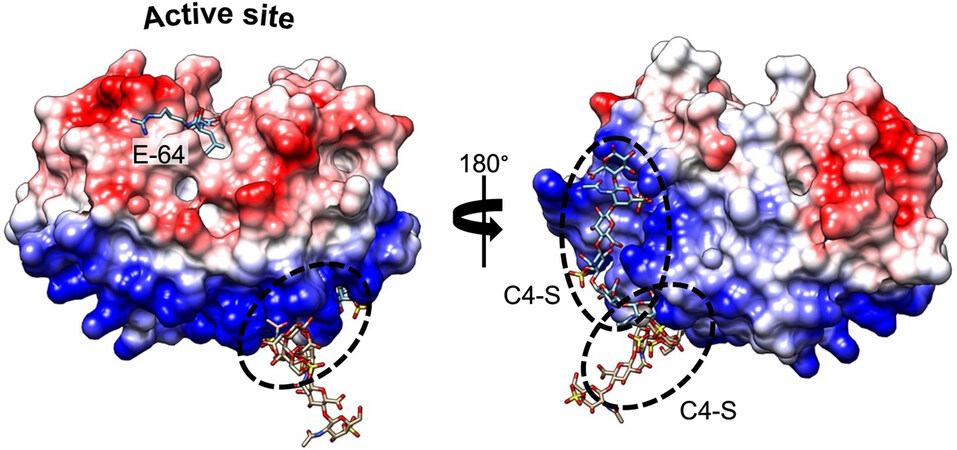
Figure 1. Complex formation between CatK and C4-S. Electrostatic surface distribution of human CatK. Blue and red areas represent positively and negatively charged surface domains of CatK, respectively (Chimera software). The spots of two C4-S-binding sites (exosites) (PDB accession codes 3C9E and 4N8W) are marked by dashed circles, away from the active site. The pan inhibitor of cysteine proteases E-64 locates in the active site of the enzyme. The peptidase is shown in surface representation, while C4-S and E-64 are shown as sticks.
Cathepsin K in osteoporosis, bone cancer and oral diseases
Besides its biological roles in bone turnover, skeletal, heart, lung and intestinal development, and reproduction, CatK expression may be dysregulated in bone resorption disorders like osteoporosis and bone metastasis[5]. CatK has been identified as the main osteoclastic bone-resorbing protease and its overexpression is markedly related to extensive bone loss, which is highlighted by the presence of N-telopeptide (NTx) collagen fragments (a typical product of CatK cleavage) in the urine/serum of osteoporotic patients[6]. Since inhibition of CatK active site could prevent bone resorption without perturbing bone formation, the protease has become an attractive and validated target for anti-resorptive drug development[34]. Moreover, abnormally high expression of CatK in various organs intertwines with the massive hydrolysis of elastin fibers or collagens in numerous cancers such as prostate, breast, lung, and bone cancers[8,10]. CatK, which contributes to tumor progression via extracellular degradation of ECM proteins, cytokines inactivation/activation or pro-matrix metalloproteinases processing, was proposed for differential prognostic purposes.
Oral and maxillofacial abnormalities were found in Ctsk(-/-) mice. Similarly, close relationships between defective CatK and oral diseases were identified in patients with pycnodysostosis[35], but also in patients with periodontitis, peri-implantitis, tooth movement, oral and maxillofacial tumor, root resorption, and periapical disease (see for review:[36]). Extensive histological and ultrastructural changes of cementum, a part of the periodontium, were observed, which might be linked to compromised proteolytic activity of CatK[37]. Otherwise, mutations of human CLCN7, which encodes voltage-gated chloride channel 7 (so-called ClC-7), lead to osteopetrosis, associated with deformities in craniofacial morphology and marked tooth dysplasia. Also, loss of CLCN7 function resulted in lysosomal storage in the brain as well as in the jaw and its surrounding, which has been associated with CatK downregulation[38].
Thus, from the X-ray crystal structure of CatK, numerous efforts have been made to develop potent, selective and orally deliverable CatK inhibitors. However, all synthetic inhibitors which have been tested in preclinical or clinical trials so far exclusively target the active site of CatK, thus blocking both its collagenolytic activity and other peptidase activities such as maturation of thyroid hormones and TGF-β1 hydrolysis. Therefore, blocking its entire active site may cause unwanted side effects [Figure 2]. Accordingly, Merck & Co had to discontinue their phase III clinical trials for osteoporosis treatment using Odanacatib, the most promising drug targeting CatK. Compared with other anti-resorptive agents, Odanacatib effectively limited osteoclast-mediated bone resorption without suppressing remodeling (preservation of bone formation). However, despite its ability to increase bone mineral density (BMD) and improve bone strength in the spine and hip in postmenopausal women with osteoporosis, Odanacatib also increases the risk of cardiovascular side effects, specifically stroke[39]. For an exhaustive update of preclinical and clinical trials conducted with CatK inhibitors, see a recent review:[34]. Thus, new pharmacological approaches are urgently needed to design novel inhibitors of human CatK to impair its collagenase activity but not other physiological regulatory proteolytic activities. One possible strategy that has emerged in recent years and could help overcome the problems associated with on-target toxicity is focusing on finding either exosite or allosteric inhibitors, such as tanshinones, which are ectosteric inhibitors isolated from the roots and rhizomes of the Chinese medicinal herb Salvia miltiorrhiza Bunge (Danshen)[40,41]. It should be noted that inhibition by gene therapy could also limit adverse off-target effects. Indeed, systemic delivery of a bone-targeting recombinant adeno-associated virus, serotype 9 (rAAV9), which can deliver to osteoclasts an artificial microRNA (rAAV9.amiR-ctsk), counteracts bone loss and improves bone mechanical properties in a murine model of postmenopausal and senile osteoporosis[42].
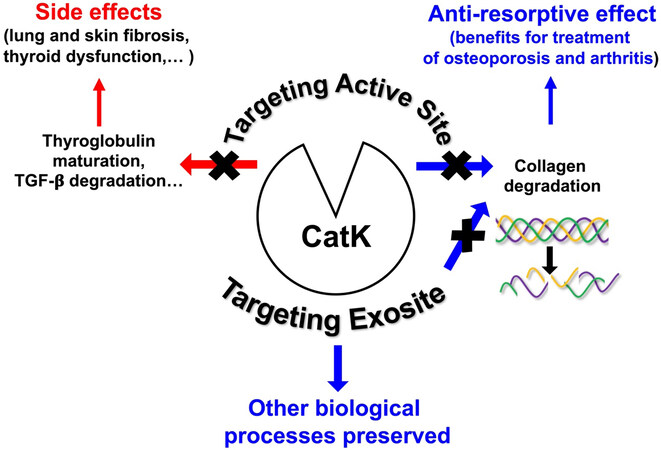
Figure 2. Selective inhibition of the collagenolytic activity of cathepsin K by targeting its exosite. Schematic drawing summarizing the overall strategy to target cathepsin K by exosite-binding inhibitors.
LYMPHANGIOLEIOMYOMATOSIS: CLINICAL FEATURES, DIAGNOSIS AND CURRENT THERAPY
Clinical features
Lymphangioleiomyomatosis (LAM) is a rare multisystemic disorder that belongs to the group of cystic pulmonary diseases and mainly affects young women. This disease frequently progresses to chronic respiratory failure. It can occur sporadically or during a genetic disease, tuberous sclerosis (also known as Bourneville’s disease)[43,44]. The prevalence of the sporadic form is estimated to be 3.4-7.8/million adult women with an incidence of 0.23 to 0.31/million women/year[45]. In France, 320 cases of patients with LAM were recorded in April 2021(RE-LAM-CE: National Register of Lymphangioleiomyomatosis). It is more common in people with tuberous sclerosis, up to 30%. This pathology affects almost exclusively women in the period of genital activity, with a median age of 35 years at the time of diagnosis. LAM is considered as a slowly progressive neoplastic disease, responsible for the proliferation of cells derived from smooth muscle cells in the lymphatic pathways, particularly in the lungs, resulting in progressive cystic lung destruction responsible for the deterioration of respiratory function[46]. The clinic is mainly marked by recurrent pneumothorax, progressively worsening dyspnea on exertion, then chronic obstructive respiratory failure. The respiratory function (i.e., Forced Expiratory Volume in the first second (FEV1) and Diffusing Capacity of the lung for carbon monoxide (DLCO) is correlated with chest Computed Tomography (CT) and histopathological abnormalities. In addition, renal angiomyolipoma injuries are observed, which can sometimes be responsible for fatal hemorrhagic events. Since the risk is correlated with the size of the tumors, regular monitoring is therefore required[47].
Diagnosis and LAM markers
Definitive diagnosis is based on tissue (most often lung) biopsy and/or the association of a clinical picture and a characteristic chest CT appearance. Histopathological diagnosis is based on the association of cystic cavities and disseminated proliferation of abnormal/ immature smooth muscle cells (LAM cells). LAM cells express smooth muscle α-actin (α-SMA), desmin, vimentin[46] and hormone receptors for estrogen and progesterone[48]. Moreover, LAM cells are characterized by their reactivity with mouse monoclonal antibody HMB45 (i.e., Human Melanoma Black), which was originally developed against human melanoma[49] and is currently used as a diagnostic marker of LAM[50]. The origin of these cells is still unknown, and the main hypothesis is uterine origin with migration via the lymphatic pathways. The mechanisms involved in the pathophysiology of LAM are misunderstood. In particular, they encompass lymphangiogenesis, which is notably associated with the overexpression of lymphatic growth factors, vascular endothelial growth factor (VEGF)-C and VEGF-D, and their receptor VEGF R3, a marker of lymphatic endothelial cells[51]. The level of blood VEGF-D, which is significantly increased in LAM, is currently the only serum biomarker for LAM diagnosis. Indeed, high levels of serum VEGF-D (> 800 pg/ml) are specifically observed in LAM, among pulmonary cystic pathologies. The pathophysiology of LAM also includes a genetic component. Mutations during tuberous sclerosis concern two tumor suppressor genes: tuberous sclerosis complex 1 (TSC1) and 2 (TSC2), coding respectively for hamartin and tuberin[52]. The TSC1/TSC2 complex allows the inhibition of the intracellular signaling pathway involved in cell growth and proliferation, the mTOR-S6k1 [mammalian target of rapamycin (mTOR)-S6 kinase 1] pathway[53]. In the case of sporadic LAM, these mutations are sometimes found at the somatic level and de novo. They are found in lung and kidney lesions, the most common mutation being that affecting the TSC2 gene. Mutations in these genes lead to a loss of inhibitory function of the hamartin-tuberin complex and are responsible for the constitutive activation of the mTOR pathway and subsequent abnormal cell proliferation and growth as well as lung remodeling [Figure 3][54]. Contrariwise, the constitutive activation of Raptor-containing mTORC1 leads to downregulation of apoptosis and cell survival[55].
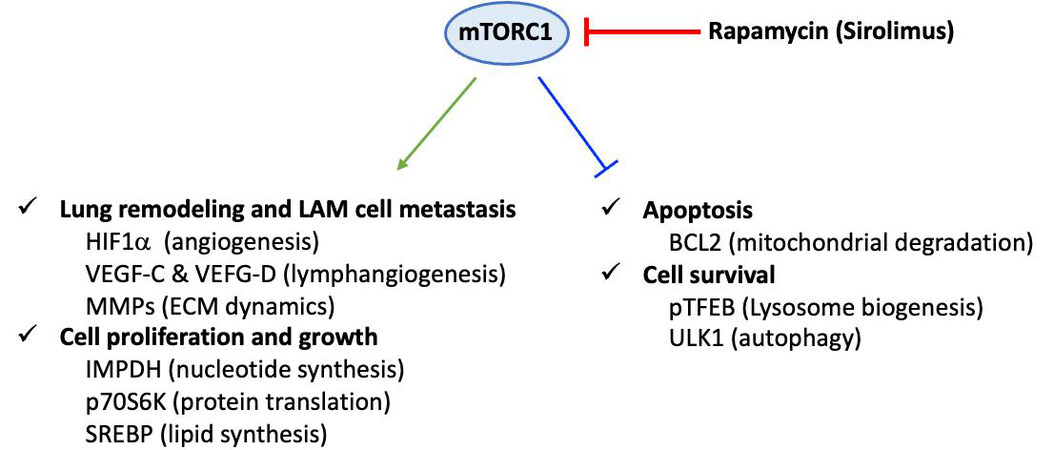
Figure 3. Dysregulation of mammalian target of rapamycin (mTOR) C1 signaling in LAM pathogenesis. Constitutive activation of the Raptor-containing mTOR leads to dysregulated mTOR signaling pathways in LAM cells. Consequently, cell proliferation, cell growth, lung remodeling as well apoptosis and cell survival are altered. This scheme corresponds to a non-exhaustive summary, emphasizing some key proteins which are specifically linked to mTOR-dependent under-expression or over-expression in LAM cells. Representative upregulated molecules: HIF-1α (hypoxia-inducible factor 1α), VEGF (vascular endothelial growth factor)-C and VEGF-D, MMP (matrix metalloproteinase), IMPDH (inosine 5'-monophosphate dehydrogenase), p70S6K (70 kDa ribosomal protein S6 kinase), SREBP (sterol regulatory element-binding protein) and downregulated molecules: BCL2 (B-cell lymphoma 2), pTFEB (phosphorylated form of transcription factor EB), ULK1 (Unc-51-like autophagy-activating kinase 1).
Current therapy
Recently, purine and pyrimidine nucleotide analogues and immune checkpoint inhibitors were advocated as potential therapeutic avenues to induce LAM cell death (see for review:[55]). Likewise, preliminary surveys of statins and a cyclo-oxygenase-2 inhibitor of the coxib family (celecoxib) were reported, but supplemental studies with larger LAM cohorts are needed to validate clinical effectiveness. Also, progesterone as well as pharmacological inhibitors (oestrogen receptor modulators, gonadotropin-releasing hormone agonists, aromatase inhibitors) were attempted as potential treatments for LAM but gave unreliable and unconvincing results[55]. Conversely, several clinical studies have pointed to a beneficial effect of mTOR inhibitors, especially rapamycin, on LAM [Figure 3]. Rapamycin (a.k.a., Sirolimus), a macrolide compound naturally produced by the bacterium Streptomyces hygroscopicus, is currently the reference treatment for this pathology[56]. Nevertheless, its effect, which remains only suspensive, is associated with many side effects, notably an increased risk of infection following the immunosuppression induced by treatment with Sirolimus[57]. Also, Sirolimus can be responsible for hematological disorders with a very frequent probability of pancytopenia associated with the risks of an infectious event[56]. Although rapamycin favors stabilization of lung function and improves quality of life, the cessation of rapamycin treatment results in recurrence of the disease progression, highlighting the imperative need to identify novel targets and contemporary LAM treatments (see for review:[58]).
CATHEPSIN K: A NEW LAM BIOMARKER?
An early immunohistochemical study reported a strong CatK immunoreactivity, which was specifically restricted to lymphangioleiomyomatosis specimen, compared to other lung samples from angiomyolipomas and diverse lung diseases (sarcoidosis, organizing pneumonia, usual interstitial pneumonia, emphysema) used as controls[59]. The specificity of CatK as a putative LAM marker seemed appropriate, according to other pulmonary α-SMA-expressing cells that are immuno-negative for CatK or exhibit only a discrete CatK immunoreactivity, such as myofibroblasts in fibroblast foci of usual interstitial pneumonia[59]. Moreover, these α-SMA-expressing cells do not represent diagnostic problems because of their distinctive morphologies and immunophenotypes. Following this study, authors proposed that CatK could be a useful additional biomarker for diagnosis in some ambiguous cases. Moreover, they suggested for the first time that CatK can contribute to the progressive remodelling of lung parenchyma observed in LAM. In an outstanding study, it was hypothesized that LAM nodule-derived proteases cause cyst formation and tissue damage[60]. A gene expression profiling analysis was performed in whole lung tissue. The authors showed that CatK gene expression was 40-fold overexpressed in LAM compared with control lung tissue, while immunohistochemistry confirmed overexpression of CatK protein in LAM tissue. Consistently, CatK immunoreactivity is predominantly co-localized with LAM-associated fibroblasts in lung nodules. Also, CatK is overexpressed in renal angiomyolipomas found in LAM, which relate to “perivascular epithelioid cell lesions” (PECome)[61]. Recently, the sensitivity of both CatK and HMB-45 were compared as potent diagnostic markers for pulmonary LAM. The percentage of LAM cells expressing CatK was significantly higher than for HMB45 and overall expression was significantly higher, confirming that CatK is a more sensitive immunohistochemical marker than HMB45 in diagnosing pulmonary LAM[62].
CATHEPSIN K: AN ADDITIONAL THERAPEUTIC TARGET IN LYMPHANGIOLEIOMYOMATOSIS?
C4-S binding is mandatory to promote the collagenolytic activity of CatK, as stated before (paragraph 1.2). C4-S is predominantly expressed in bone and cartilage, but is also found throughout the body and contributes to ECM remodeling processes in numerous chronic inflammatory diseases[63]. Although the expression level of C4-S is currently not known in LAM, the total amount of GAG is increased in lung diseases, including idiopathic pulmonary fibrosis (IPF) or mucopolysaccharidosis (MPS), another rare disease[63,64]. Alongside its immunoreactivity, active CatK was detected in LAM-associated fibroblasts. Also, active secreted CatK was found in extracellular medium under in vitro conditions. It is well established that monocyte-derived macrophages acidify their pericellular environment via vacuolar-type H+-ATPases, thus enabling them to maintain cysteine cathepsins, including CatK, in their active form[21]. Similar extracellular acidification may exist within LAM nodules, because of both expression of membrane transporters (carbonic anhydrases, monocarboxylate transporters and sodium-bicarbonate co-transporters) and mTOR dysregulation, which induces a metabolic dependence on aerobic glycolysis (Warburg effect). Acidification paralleled CatK activity, and both were forcefully compromised by sodium bicarbonate co-transporter inhibitors or carbonic anhydrase inhibitors. Impressively, inhibition of mTOR by rapamycin also abrogated acidification in a cell culture model by acting on cell metabolism rather than solely on the transporters themselves. Thus, inhibition of extracellular acidification may be an alternative therapy for LAM by indirect impairment of CatK activity[60].
As previously mentioned, inhibition of the mTOR pathway by rapamycin is currently the only reference therapy for LAM. This beneficial effect may be related to the suppression of Warburg metabolism and extracellular acidification. Accordingly, pharmacological inhibitors of carbonic anhydrases and sodium-bicarbonate co-transporters have been successfully used in preclinical cancer models[65,66] and may have synergistic benefits with mTOR inhibition in LAM to reduce detrimental proteases activity, including CatK. Interestingly, markers for LAM cells, fibroblasts, lymphatics, and mast cells were examined by dual immunohistochemistry, quantitated in LAM nodules, and compared with clinical features and prospective lung function loss. Levels of CatK were negatively correlated with FEV1 and DLCO, while a higher reactivity to the mTOR complex 1 activation marker was associated with a better lung function response to rapamycin[67]. Taken together, results suggested that an increase in CatK expression is associated with severity of the disease and loss of lung function. At last, the higher prevalence of osteoporosis in LAM than in the general population strengthens the interest in specifically targeting CatK[68]. Interestingly, some recent reports suggested that overexpression of CatK is associated with mTOR upregulation[69], while CatK could promote tumor cell proliferation, invasion and migration, and its mechanism may be related to mTOR signaling pathway[70]. On the other hand, expression of cysteine cathepsins (including CatK and CatS) is associated, via the inhibition of mTOR, with a decreased phosphorylation of TFEB (transcription factor EB), a central regulator of autography/lysosomal biogenesis[34,71]. Therefore, although there is a scarcity of experimental data, the possibility that rapamycin may have an impact on CatK expression cannot be ruled out.
To summarize, combining the current treatment with rapamycin with the inhibition of collagenolytic CatK by ectosteric inhibitors could be more effective than mTOR inhibition alone. Indeed, a cooperative curative outcome in reducing lung destruction in LAM could be expected compared to a single therapeutic targeting. In our opinion, such a dual approach is conceivable despite a lack of experimental evidence at this hypothetical clinical stage.
DECLARATIONS
AcknowledgmentsWe gratefully acknowledge the patient association “French Lymphangioleiomyomatosis” (FLAM) for their kind support. We thank The University Hospital Center of Tours (CHU Tours, Protocol Collaboration Agreement: RIPH3 / LAM-CAK).
Authors’ contributionsDraft the paper: Lalmanach G, Lecaille F, Marchand-Adam S
Prepared the figures: Lecaille F, Lalmanach G
Wrote the paper: Lalmanach G
Revised the paper: Saidi A, Pronost M
All authors read and approved the final manuscript
Availability of data and materialsNot applicable.
Financial support and sponsorshipNot applicable.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Lecaille F, Kaleta J, Brömme D. Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem Rev 2002;102:4459-88.
2. Turk V, Stoka V, Vasiljeva O, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012;1824:68-88.
3. Brix K, Dunkhorst A, Mayer K, Jordans S. Cysteine cathepsins: cellular roadmap to different functions. Biochimie 2008;90:194-207.
4. Kramer L, Turk D, Turk B. The future of cysteine cathepsins in disease management. Trends Pharmacol Sci 2017;38:873-98.
5. Lecaille F, Brömme D, Lalmanach G. Biochemical properties and regulation of cathepsin K activity. Biochimie 2008;90:208-26.
6. Brömme D, Lecaille F. Cathepsin K inhibitors for osteoporosis and potential off-target effects. Expert Opin Investig Drugs 2009;18:585-600.
7. Novinec M, Lenarčič B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem 2013;394:1163-79.
8. Qian D, He L, Zhang Q, et al. Cathepsin K: a versatile potential biomarker and therapeutic target for various cancers. Curr Oncol 2022;29:5963-87.
9. Rocho FR, Bonatto V, Lameiro RF, Lameira J, Leitão A, Montanari CA. A patent review on cathepsin K inhibitors to treat osteoporosis (2011-2021). Expert Opin Ther Pat 2022;32:561-73.
10. Verbovšek U, Van Noorden CJ, Lah TT. Complexity of cancer protease biology: cathepsin K expression and function in cancer progression. Semin Cancer Biol 2015;35:71-84.
12. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 1967;27:157-62.
13. Jaffer FA, Kim DE, Quinti L, et al. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation 2007;115:2292-8.
14. Lecaille F, Weidauer E, Juliano MA, Brömme D, Lalmanach G. Probing cathepsin K activity with a selective substrate spanning its active site. Biochem J 2003;375:307-12.
15. Lemke C, Benýšek J, Brajtenbach D, et al. An activity-based probe for cathepsin K imaging with excellent potency and selectivity. J Med Chem 2021;64:13793-806.
16. Richard ET, Morinaga K, Zheng Y, et al. Design and synthesis of cathepsin-k-activated osteoadsorptive fluorogenic sentinel (OFS) probes for detecting early osteoclastic bone resorption in a multiple myeloma mouse model. Bioconjug Chem 2021;32:916-27.
17. Li Z, Hou WS, Brömme D. Collagenolytic activity of cathepsin K is specifically modulated by cartilage-resident chondroitin sulfates. Biochemistry 2000;39:529-36.
18. Li Z, Hou W-S, Escalante-Torres CR, et al. Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J Biol Chem 2002;277:28669-28676.
19. Fonović M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta 2014;1840:2560-70.
20. Brisson L, Reshkin SJ, Goré J, Roger S. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol 2012;91:847-60.
21. Punturieri A, Filippov S, Allen E, et al. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin K-deficient human macrophages. J Exp Med 2000;192:789-99.
22. Brömme D, Okamoto K. Human cathepsin O2, a novel cysteine protease highly expressed in osteoclastomas and ovary molecular cloning, sequencing and tissue distribution. Biol Chem Hoppe Seyler 1995;376:379-384.
23. Quintanilla-Dieck MJ, Codriansky K, Keady M, Bhawan J, Rünger TM. Expression and regulation of cathepsin K in skin fibroblasts. Exp Dermatol 2009;18:596-602.
24. Friedrichs B, Tepel C, Reinheckel T, et al. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest 2003;111:1733-45.
25. Tepel C, Brömme D, Herzog V, Brix K. Cathepsin K in thyroid epithelial cells: sequence, localization and possible function in extracellular proteolysis of thyroglobulin. J Cell Sci 2000;113 Pt 24:4487-98.
26. Bühling F, Waldburg N, Reisenauer A, et al. Lysosomal cysteine proteases in the lung: role in protein processing and immunoregulation. Eur Respir J 2004;23:620-628.
27. Zhang D, Leung N, Weber E, Saftig P, Brömme D. The effect of cathepsin K deficiency on airway development and TGF-β1 degradation. Respir Res 2011;12:72.
29. Chen H, Wang J, Xiang MX, et al. Cathepsin S-mediated fibroblast trans-differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovasc Res 2013;100:84-94.
30. Yue X, Piao L, Wang H, et al. Cathepsin K deficiency prevented kidney damage and dysfunction in response to 5/6 nephrectomy injury in mice with or without chronic stress. Hypertension 2022;79:1713-23.
31. Zhang X, Zhou Y, Yu X, et al. Differential roles of cysteinyl cathepsins in tgf-β signaling and tissue fibrosis. iScience 2019;19:607-22.
32. Lalmanach G, Saidi A, Marchand-Adam S, Lecaille F, Kasabova M. Cysteine cathepsins and cystatins: from ancillary tasks to prominent status in lung diseases. Biol Chem 2015;396:111-30.
33. Kasabova M, Joulin-Giet A, Lecaille F, et al. Regulation of TGF-β1-driven differentiation of human lung fibroblasts: emerging roles of cathepsin B and cystatin C. J Biol Chem 2014;289:16239-51.
34. Biasizzo M, Javoršek U, Vidak E, Zarić M, Turk B. Cysteine cathepsins: a long and winding road towards clinics. Mol Aspects Med 2022;88:101150.
35. Henriksen K, Thudium CS, Christiansen C, Karsdal MA. Novel targets for the prevention of osteoporosis - lessons learned from studies of metabolic bone disorders. Expert Opin Ther Targets 2015;19:1575-84.
36. Wen X, Yi LZ, Liu F, Wei JH, Xue Y. The role of cathepsin K in oral and maxillofacial disorders. Oral Dis 2016;22:109-15.
37. Xue Y, Wang L, Xia D, et al. Dental abnormalities caused by novel compound heterozygous
38. Zhang Y, Ji D, Li L, Yang S, Zhang H, Duan X. ClC-7 Regulates the pattern and early development of craniofacial bone and tooth. Theranostics 2019;9:1387-400.
39. McClung MR, O'Donoghue ML, Papapoulos SE, et al. LOFT Investigators. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol 2019;7:899-911.
40. Panwar P, Law S, Jamroz A, et al. Tanshinones that selectively block the collagenase activity of cathepsin K provide a novel class of ectosteric antiresorptive agents for bone. Br J Pharmacol 2018;175:902-23.
41. Panwar P, Xue L, Søe K, et al. An ectosteric inhibitor of cathepsin K inhibits bone resorption in ovariectomized mice. J Bone Miner Res 2017;32:2415-30.
42. Yang YS, Xie J, Chaugule S, et al. Bone-Targeting AAV-mediated gene silencing in osteoclasts for osteoporosis therapy. Mol Ther Methods Clin Dev 2020;17:922-35.
43. Cottin V, Archer F, Khouatra C, Lazor R, Cordier JF. Lymphangioléiomyomatose pulmonaireLymphangioleiomyomatosis. Presse Med 2010;39:116-25.
44. Johnson SR, Cordier JF, Lazor R, et al. Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010;35:14-26.
45. Harknett EC, Chang WY, Byrnes S, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM 2011;104:971-9.
46. Henske EP, McCormack FX. Lymphangioleiomyomatosis - a wolf in sheep’s clothing. J Clin Invest 2012;122:3807-16.
47. Taveira-DaSilva AM, Moss J. Epidemiology, pathogenesis and diagnosis of lymphangioleiomyomatosis. Expert Opin Orphan Drugs 2016;4:369-78.
48. Ruiz de Garibay G, Herranz C, Llorente A, et al. Lymphangioleiomyomatosis biomarkers linked to lung metastatic potential and cell stemness. PLoS One 2015;10:e0132546.
49. Gown AM, Vogel AM, Hoak D, et al. Monoclonal antibodies specific for melanocytic tumors distinguish subpopulations of melanocytes. Am J Pathol 1986;123:195-203.
50. Matsumoto Y, Horiba K, Usuki J, Chu SC, Ferrans VJ, Moss J. Markers of cell proliferation and expression of melanosomal antigen in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol 1999;21:327-36.
51. Young L, Lee HS, Inoue Y, et al. MILES Trial Group. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med 2013;1:445-52.
52. Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet 2018;178:326-37.
53. Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet 1998;62:810-5.
54. Yu J, Henske EP. mTOR activation, lymphangiogenesis, and estrogen-mediated cell survival: the “perfect storm” of pro-metastatic factors in LAM pathogenesis. Lymphat Res Biol 2010;8:43-9.
55. McCarthy C, Gupta N, Johnson SR, Yu JJ, McCormack FX. Lymphangioleiomyomatosis: pathogenesis, clinical features, diagnosis, and management. Lancet Respir Med 2021;9:1313-27.
56. Harari S, Spagnolo P, Cocconcelli E, Luisi F, Cottin V. Recent advances in the pathobiology and clinical management of lymphangioleiomyomatosis. Curr Opin Pulm Med 2018;24:469-76.
57. Courtwright AM, Goldberg HJ, Henske EP, El-Chemaly S. The effect of mTOR inhibitors on respiratory infections in lymphangioleiomyomatosis. Eur Respir Rev 2017;26:160004.
58. Moir LM. Lymphangioleiomyomatosis: Current understanding and potential treatments. Pharmacol Ther 2016;158:114-24.
59. Chilosi M, Pea M, Martignoni G, et al. Cathepsin-k expression in pulmonary lymphangioleiomyomatosis. Mod Pathol 2009;22:161-166.
60. Dongre A, Clements D, Fisher AJ, Johnson SR. Cathepsin K in lymphangioleiomyomatosis: LAM cell-fibroblast interactions enhance protease activity by extracellular acidification. Am J Pathol 2017;187:1750-62.
61. Caliò A, Brunelli M, Gobbo S, et al. Cathepsin K: a novel diagnostic and predictive biomarker for renal tumors. Cancers (Basel) 2021;13:2441.
62. Rolim I, Makupson M, Lovrenski A, Farver C. Cathepsin K is superior to HMB45 for the diagnosis of pulmonary lymphangioleiomyomatosis. Appl Immunohistochem Mol Morphol 2022;30:108-12.
63. Westergren-Thorsson G, Hedström U, Nybom A, et al. Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2017;83:27-38.
64. Chazeirat T, Denamur S, Bojarski KK, et al. The abnormal accumulation of heparan sulfate in patients with mucopolysaccharidosis prevents the elastolytic activity of cathepsin V. Carbohydr Polym 2021;253:117261.
65. Kant S, Kumar A, Singh SM. Bicarbonate transport inhibitor SITS modulates pH homeostasis triggering apoptosis of Dalton’s lymphoma: implication of novel molecular mechanisms. Mol Cell Biochem 2014;397:167-78.
66. Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896-902.
67. Miller S, Stewart ID, Clements D, Soomro I, Babaei-Jadidi R, Johnson SR. Evolution of lung pathology in lymphangioleiomyomatosis: associations with disease course and treatment response. J Pathol Clin Res 2020;6:215-26.
68. Taveira-Dasilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Bone mineral density in lymphangioleiomyomatosis. Am J Respir Crit Care Med 2005;171:61-7.
69. Iakymenko OA, Delma KS, Jorda M, Kryvenko ON. Cathepsin K (Clone EPR19992) demonstrates uniformly positive immunoreactivity in renal oncocytoma, chromophobe renal cell carcinoma, and distal tubules. Int J Surg Pathol 2021;29:600-5.
70. Seo SU, Woo SM, Kim MW, et al. Cathepsin K inhibition-induced mitochondrial ROS enhances sensitivity of cancer cells to anti-cancer drugs through USP27x-mediated Bim protein stabilization. Redox Biol 2020;30:101422.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Marchand-Adam S, Pronost M, Saidi A, Lecaille F, Lalmanach G. Cathepsin K: both a likely biomarker and a new therapeutic target in lymphangioleiomyomatosis?. Rare Dis Orphan Drugs J 2023;2:3. http://dx.doi.org/10.20517/rdodj.2022.24
AMA Style
Marchand-Adam S, Pronost M, Saidi A, Lecaille F, Lalmanach G. Cathepsin K: both a likely biomarker and a new therapeutic target in lymphangioleiomyomatosis?. Rare Disease and Orphan Drugs Journal. 2023; 2(1): 3. http://dx.doi.org/10.20517/rdodj.2022.24
Chicago/Turabian Style
Marchand-Adam, Sylvain, Marion Pronost, Ahlame Saidi, Fabien Lecaille, Gilles Lalmanach. 2023. "Cathepsin K: both a likely biomarker and a new therapeutic target in lymphangioleiomyomatosis?" Rare Disease and Orphan Drugs Journal. 2, no.1: 3. http://dx.doi.org/10.20517/rdodj.2022.24
ACS Style
Marchand-Adam, S.; Pronost M.; Saidi A.; Lecaille F.; Lalmanach G. Cathepsin K: both a likely biomarker and a new therapeutic target in lymphangioleiomyomatosis?. Rare. Dis. Orphan. Drugs. J. 2023, 2, 3. http://dx.doi.org/10.20517/rdodj.2022.24
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 34
likes
Like This Article 34
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.