
Immune cell-derived serine protease as pathogenic drivers of vascular remodeling in pulmonary arterial hypertension
 0
0 ,
, Abstract
In recent years, accumulating evidence has shown that pulmonary arterial hypertension (PAH) has a strong underlying inflammatory component. Vascular remodeling, a common pathology observed in all forms of pulmonary hypertension (PH), is accompanied by a pronounced accumulation of leukocytes around and within the vessels. Proteolytic products of immune cells, particularly neutrophil and mast cell serine proteases, have been shown to play a central pathogenic role in vascular remodeling and PAH development. Serine proteases are involved in many aspects of the inflammatory response, such as extracellular matrix degradation, regulation of bioavailability of cytokines, chemokines, and growth factors, and dysregulation of their activity can have devastating consequences. In this review, we will focus on immune dysregulation in PAH and shed light on the pro-inflammatory role of serine proteases in vascular pathology observed in the context of this disease.
Keywords
PULMONARY ARTERIAL HYPERTENSION
Pulmonary arterial hypertension (PAH) is a severe clinical condition characterized by persistent elevation of pulmonary arterial pressure (> 20 mmHg)[1]. Even with the best standard of care, the overall prognosis for PAH patients remains poor, with a 3-year mortality rate of 21%. The picture looks even worse for patients from the high-risk PAH group with a 3-year mortality rate of 28%-55%[2]. PH can develop as a primary disease with an unknown cause (idiopathic, IPAH), as a result of the loss-of-function mutation in the gene encoding the bone morphogenetic protein receptor type II (BMPR-2, heritable PAH), or in association with other diseases[3]. However, irrespective of the underlying cause, all pulmonary hypertension forms are accompanied by progressive vascular remodeling[4], evident in small and medium size distal pulmonary arteries (PAs). Narrowing of the vessel lumen, together with reduced vasodilatory capacity, results in an increased pulmonary vascular resistance (PVR), progressively leading to right ventricle (RV) hypertrophy, heart failure, and ultimately premature death[5]. Pathogenesis of PAH is very complex and multifactorial and consensus on what triggers the disease development is missing. Affected vessels show pulmonary arterial endothelial cell (PAEC) dysfunction, expansion of media caused by abnormal proliferation of pulmonary arterial smooth muscle cells (PASMC), and changes in the extracellular matrix (ECM) architecture with collagen deposition[6] and elastic lamina breakdown[7]. Vessel remodeling is commonly accompanied by perivascular inflammation and cumulative evidence suggests that active recruitment of immune cells and inflammatory mediators, such as serine proteases, can drive pathogenic processes in PAH[8-10]. In this review, we will focus on the role of aberrant inflammatory response and serine proteases-mediated vascular injury in the initiation and propagation of PH-associated vasculopathy. We will also briefly discuss the utility of serine protease targeting as a novel immunomodulatory, anti-remodeling treatment strategy in PH.
Immune dysregulation in PH
There is growing evidence that maladaptive immune response plays an important role in PAH pathogenesis. The presence of several types of autoantibodies against vascular wall components has been identified in PAH[11,12]. Inflammatory mediators, involved in the leukocyte recruitment and activation, are elevated in the circulation and lungs of patients, and their increased levels correlate with worse clinical outcomes (e.g., interleukins IL-1β, IL-2, IL-4, IL-6, IL-8, MCP1, CCL5, CX3CL1, and TNFα)[13-16]. Furthermore, an increased prevalence of PH is observed in individuals suffering from diseases associated with maladaptive immune responses, e.g., scleroderma, systemic lupus erythematosus, sarcoidosis, and chronic obstructive pulmonary disease (COPD)[17-20]. In experimental PAH, inflammation proceeds the development of vascular changes, suggesting its causal role in vascular remodeling[21,22]. Interestingly, single-cell interaction mapping identified the remarkable breakdown of intracellular communication in diseased vessels. While in healthy PAs, harmonious crosstalk between the immune and structural cellular compartments can be observed, in PAH, there is a strong upregulation of ligand-receptor pairs between PASMC and granulocytes[23]. This indicates that in PAH, vascular cells enforce communication both within the vessel and with the granulocyte compartment.
Endothelial dysfunction is believed to be one of the triggers initiating a detrimental process that ultimately leads to vascular remodeling and evidence of an activated pulmonary endothelium is commonly found in PAH patients[24]. Increased expression of leukocyte adhesion molecules and inflammatory mediators by endothelial cells provides a perfect gateway for the tissue accumulation of inflammatory leukocytes[25]. Immune cell aggregates appear to be associated with active arterial remodeling, and perivascular inflammation score correlates with PA hyperplasia, pulmonary hemodynamics, and clinical outcome[9]. The role of specific immune cell types has been investigated in the context of PAH pathogenesis. However, only recently has it been shown in the systematic analysis of human samples that in comparison to healthy donors, IPAH patients have markedly changed global immune landscape both in the lung parenchyma and the PAs[23,26]. A multitude of different cells implicated in the disease process in PAH underpins the complexity of the innate and adaptive immune cell crosstalk in disease development. Already in the late '60s, the association of mast cells (MCs) with remodeled vessels has been shown[27]. Notably, the depletion of MCs in experimental PAH, as well as inhibition of their activity, attenuates pulmonary vascular remodeling[28-31]. Regulatory T cells (Tregs), natural killer (NK) cells, and NKT cells appear to play a beneficial role in the maintenance of vascular homeostasis and their deficiency and altered function have been linked to PA remodeling and fibrosis[32-35]. Experimental and clinical PAH is also accompanied by increased numbers and activation of dendritic cells (DCs) and macrophages in remodeled vessels[10,36,37]. The development of inflammatory macrophages depends on the influx of monocytes from the circulation and analysis of lung tissue from hypoxic mice and patients with PAH shows increased levels of factors that promote monocyte migration (e.g., CCL1, CCL2, CX3CL1)[38,39]. Moreover, the lungs of IPAH patients show evidence of lymphoid neogenesis. Adjacent to remodeled vessels, organized tertiary lymphoid tissues (TLT), comprised of B and T cell zones, can be found, raising an intriguing possibility of local adaptive immune response in these patients[10]. Growing attention is given to the role of neutrophils in PAH pathogenesis. Increased neutrophil to lymphocyte ratio (NLR) has been identified in the blood of the patients and NLR correlates with prognostic PAH biomarkers. Higher NLR is associated with increased levels of N-terminal pro-brain natriuretic peptide (NT-proBNP), rise in right atrial pressure (RAP), and shorter 6-minute walking distance (6MWD)[40,41]. Furthermore, elevated NLR is linked to increased morbidity, mortality, and unfavorable transplantation-free survival, independent of hemodynamic parameters and C-reactive protein[41]. Activated neutrophils can extrude their intracellular content of chromatin fibers and granule-associated proteases, forming net-like structures called neutrophil extracellular traps (NETs). Under physiological circumstances, this process (NETosis) plays a fundamental role in pathogen clearance[42]. However, excessive NETosis has been linked to vascular damage and inflammation[43]. Elevated levels of NETosis markers, i.e., DNA-myeloperoxidase complexes, neutrophil elastase, and citrullinated histone H3, can be found both in the circulation and occlusive vascular lesions in the lungs of PAH patients[44].
Cumulatively, a large body of evidence supports the notion that vessel remodeling in PAH is accompanied by an accumulation of several different types of leukocytes which mediate a broad spectrum of pro-inflammatory actions in affected vasculature and surrounding lung tissue.
IMMUNE CELL-DERIVED SERINE PROTEASES ARE IMPORTANT REGULATORS OF INFLAMMATION AND IMMUNITY
Immune cells are a major source of proteolytic activity in the lung. Serine proteases, abundantly expressed by different types of immune cells, are involved in the processes related to immunity and inflammation in various ways (reviewed by Korkmaz, et al.[45]). They can directly degrade ECM components (e.g., collagen, elastin, and fibronectin), interact with protease-activated receptors (PARs), and regulate the availability and activity of cytokines, chemokines, and growth factors[46]. Three main serine proteases subgroups, classified by their specific interaction with a substrate, include: trypsin-like serine proteases (e.g., tryptases and granzymes), chymotrypsin-like serine proteases (e.g., chymases), and elastases (e.g., neutrophil elastase and proteinase 3). Given the potentially destructive nature of serine proteases, it is not surprising that their activity is tightly regulated through various mechanisms. Genes encoding neutrophil granule-associated serine proteases are highly expressed only during early myelopoiesis[47]. Furthermore, many serine proteases are produced as inactive pro-forms (zymogens) and require additional post-translational modification steps, such as cleavage of the N- and C-terminal domains, to become fully active. The maturation of NSPs is mediated by other enzymes, e.g., cathepsin C (CtsC), as they do not possess autocatalytic properties[48]. Immune cells retain and store serine proteases in intracellular granules bound to proteoglycans. This modulates their activity, limits access to cellular targets, and prevents extracellular leakage. Transcriptional timing determines which proteases are packaged into specific classes of granules[47]. Additionally, serine protease activity is regulated by specialized inhibitors, with serpins representing the largest and most diverse family, consisting of over 1,000 identified members across species. Serpins mimic potential substrates and form irreversible, stable complexes with proteases through a suicide mechanism, leading to loss of catalytic function. Other endogenous inhibitors, such as secretory leukocyte protease inhibitor (SLPI), elafin, and α-macroglobulins, provide an additional layer of regulation[45]. The multilevel regulation of serine protease activity ensures a balance between proteases and their inhibitory mechanisms, which is necessary for the maintenance of homeostatic equilibrium and tissue integrity. In the next part, we will discuss how inadequate control of serine proteases can be a central component of the pathogenesis of vascular remodeling in PAH.
NEUTROPHIL SERINE PROTEASES IN PAH
Neutrophil serine proteases (NSPs) are a class of granule-associated enzymes essential for intracellular pathogen killing. However, upon cell activation NSPs can also be released into extracellular space where they degrade ECM components and activate inflammatory mediators. The role of NSPs in the inflammatory response is supported by the observations made in transgenic mice. Animals that lack either NSPs or the enzyme that regulates their maturation, i.e., cathepsin C, show an impaired inflammatory response[48,49]. The most abundant serine proteases in neutrophil granules are neutrophil elastase (NE), cathepsin G (CatG), and proteinase 3 (PR3). Although NSPs are produced primarily by neutrophils, it is important to note that other immune cells, including B cells, monocytes, macrophages, and mast cells, can also produce these enzymes[45].
Neutrophil elastase (NE)
Neutrophils are a dominant source of NE. However, expression of this serine protease can also be detected in structural cells, e.g smooth muscle cells[50] and endothelial cells[51]. The increased neutrophil-to-leukocyte ratio is commonly found in PAH patients[41]. The observed difference in neutrophils is not only quantitative but also qualitative. When compared to the healthy controls, neutrophils isolated from the blood of PAH patients respond to stimulation with enhanced release of NE[52]. A recent observational study in the PAH cohort confirmed that increased serum levels of NE, with a concomitant deficiency in NE inhibitor (elafin), can be found across PAH subtypes[53]. Furthermore, a high NE level has a prognostic value and is associated with a worse clinical outcome, making circulating NE a promising PAH biomarker[53]. NE is thought to contribute to vessel remodeling in several ways. First, neointimal lesions show altered ECM composition and fragmented elastic lamina[7,54]. Elastolytic activity within the vessel generates elastin peptides and releases ECM-bound growth factors. These products can provide a chemotactic signal for the recruitment of immune cells and stimulate the proliferation and migration of PASMCs[55,56]. NE can contribute to remodeling not only by targeting elastin; it also cleaves many other ECM proteins, i.e., collagens, fibronectin, and laminin[57], and activates different proteolytic enzymes, e.g., matrix metalloproteinases (MMPs), which potentiate matrix degradation and remodeling[58]. Furthermore, neutrophil proteases propagate inflammatory signals via the modulation of the bioactivity of cytokines and chemokines[59]. NSPs are crucial components of the antimicrobial response. They are capable of effectively killing bacteria, but this occurs typically in the micromolar concantration commonly found within phagolysosomes. However, the concentration of proteases released during the degranulation of activated neutrophils is likely much lower[9,60,61]. It is noteworthy that even at very low concentrations, neutrophil-derived proteases can activate the IL-1 cytokine family, indicating that proteases released during degranulation have an important role in regulating the inflammatory response rather than serving primarily as effectors in microbial killing[61].
Inflammatory tissue injury starts with the adhesion of neutrophils to vascular intima, and the association between endothelial dysfunction, characterized by altered vasodilatory capacity, cell survival and growth factor production, and PAH is firmly established[24]. NE has been shown to damage the protective glycocalyx layer on endothelial cells, leading to endothelial injury and subsequently increased retention of leukocytes in the vessel[62-64]. Elastase can also directly affect PAECs function and induce apoptosis and endothelial barrier disruption[53,65,66]. In the vasculature, bone morphogenetic protein receptor type 2 (BMPR-2) plays an important role in the suppression of inflammation and supports the maintenance of the endothelial barrier function[67]. Enzymatic activity of NE has been shown to downregulate BMPR-2 signaling and thus could be involved in the perpetuation of inflammatory response in the vessels[68].
Several studies point to the utility of NE activity inhibition as a promising treatment strategy in PAH. Transgenic animals with overexpression of NE inhibitor, elafin, are protected from chronic hypoxia-induced PH. Compared to the control animals, they show improved hemodynamics and attenuated vascular remodeling[69]. Pharmacological inhibition of NE shows the same beneficial effects in different experimental models of PH[50,68,70]. Evaluation of Elafin in the randomized, placebo-controlled, blinded, phase I clinical trial is currently ongoing (ClinicalTrials.gov Identifier: NCT03522935).
Proteinase 3 (PR3)
PR3 is another granule-associated serine proteinase involved in neutrophilic inflammation. It has high sequence homology to NE and exhibits similar substrate specificity. Like NE, PR3 can hydrolyze elastin and other important ECM components. However, PR3 also has some unique properties, e.g., a substantial amount of this protease is present on the neutrophil surface, bound to the cell membrane. Membrane association increases even further in activated neutrophils[71]. The role of PR3 in the pathogenesis of PAH has never been specifically addressed. However, indirect evidence supports its involvement in the disease process. In particular, PR3 can efficiently convert TNFα and IL1β to their bioactive forms[72] and TNFα has been shown to be a central driver of PAH. Out of several cytokines implicated in PAH, TNFα selectively reduces the expression of BMPR-2 in PASMCS and PAECs[73], and deficiency of the BMPR-2 is a genetic risk factor for the development of the hereditary form of PH[74]. TNFα-induced alteration of BMP signaling results in excessive proliferation of PASMCs[73], thus contributing to vascular hyperplasia. Of note, the therapeutic effect of elafin in PAH could be reinforced by its ability to modulate also PR3 activity[75,76].
Cathepsin G (CatG)
CatG shares its primary specificity with NE and PR3, and it is an essential component of neutrophil granule-associated proteolytic machinery involved in a broad range of neutrophil functions. As in the case of PR3, the role of CatG in PAH has never been directly addressed. However, many processes that play a role in PAH pathogenesis, e.g., regulation of vascular tone, different aspects of the inflammatory response, and tissue remodeling, are influenced by CatG activity.
CatG and NE can convert pro-chemerin into its bioactive form[77]. There is growing evidence to suggest that chemerin plays an important role in regulating energy metabolism, cardiovascular homeostasis, and inflammatory response (reviewed by Macvanin MT, et al.[78]). Chemerin can induce the chemotaxis of dendritic cells and macrophages[79] and modulate angiogenesis[78]. Notably, vascular cells express the chemerin receptor, chemokine-like receptor 1 (CMKLR1), and Chemerin-CMKLR1 signaling has been shown to contribute to endothelial dysfunction, increase apoptosis resistance, and induce proliferation and motility of PASMCs[78,80]. Additionally, chemerin has been shown to potentiate the vasoconstrictive and mitogenic action of endothelin 1[81]. PAH patients have increased serum levels of both chemerin[82] and endothelin 1[83], possibly contributing to their commonly observed chronic vasoconstrictive state. Furthermore, neutrophil membrane-bound Cathepsin G has a catalytic activity that can generate the vasoactive hormone angiotensin II, a central component of the renin-angiotensin-aldosterone system (RAAS) responsible for regulating blood pressure[84]. Angiotensin II promotes vasoconstriction and stimulates aldosterone release, which activates kidney sodium retention, leading to elevated blood pressure. PAH patients show increased serum levels of angiotensin II as well as elevation of the RAAS activity in the pulmonary vasculature, contributing to vasoconstriction, vascular remodeling, and disease development[85].
Thromboembolic lesions are another significant pathology commonly found in the pulmonary vasculature of PAH patients[86]. CatG-mediated activation of protease-activated receptor 4 (PAR4) on platelets stimulates their aggregation, granule secretion, and leukocyte interaction[87,88], thereby contributing to the thrombo-inflammatory process in the vessels. Furthermore, CatG is involved in chemotaxis regulation. It generates active forms of CXCL5 and CCL15, thus providing a signal for the accumulation of neutrophils and monocytes, respectively[89,90]. Additionally, similar to other cathepsins, CatG is also involved in proteolytic antigen processing by antigen-presenting cells[91].
MAST CELL SERINE PROTEASES IN PAH
Neutrophil NSPs are not the only serine proteases implicated in PAH. There is also a body of evidence that links mast cell (MCs) proteolytic activity to the pathogenic process in this disease. Indeed, accumulation of MCs is commonly observed in and around the remodeled vessel[31,92] and their depletion[28], as well as inhibition of MC degranulation, ameliorates experimental PH[28,93]. Mast cells are a tissue-resident, long-lived granulocyte population that resides at the host-external environment interface, often in proximity to blood vessels. They play an important homeostatic role, coordinating inflammatory and repair processes in the tissue. Despite being very rare, the biological response evoked by MCs products can be extremely potent. Inappropriate MC activity contributes to acute allergic responses, chronic inflammation, and tissue remodeling and has been linked not only to pathology in PAH but also to other chronic pulmonary diseases, e.g., asthma, pulmonary fibrosis, and COPD[94]. Proteases can make up over 25% of total protein content in MCs, with mRNA levels that exceed commonly used housekeeping genes[95-97]. Two of the most abundant MC serine proteases, tryptase and chymase[98], have been reported to be increased in PAH[92,93]. Higher levels of circulating MC proteases, and expansion of tryptase and chymase-positive cells in the lung, correlate with the severity of the pulmonary vascular disease[31,92,99].
Tryptase
Tryptase can activate protease-activated receptor 2 (PAR2), which increased expression has been found in PASMCs in vascular lesions. The tryptase-PAR2 axis has been shown to regulate several cellular effects central to PAH pathogenesis. Primary PASMCs exposed to tryptase increase their proliferation, migration, and production of fibronectin and MMP-2[31].
Tryptase can also alter the behavior of endothelial cells and stimulate them to release neutrophil-chemotactic IL-8[100,101], and degrade collagen IV, which is a major constituent of the pulmonary vessels’ basement membrane, important for endothelial barrier function[54,102]. Furthermore, deposition of collagen is commonly observed within remodeled vessels[6,35]. Tryptase activity could facilitate this process via its ability to stimulate fibroblast proliferation, migration, and collagen I production[103-105]. In conclusion, tryptase exerts variable effects on different cell types, which cumulatively can contribute to vessel remodeling.
Chymase
Chymase, similar to CatG, has chymotrypsin-like activity. Chymase can directly (independently of angiotensin-converting enzymes) mediate local production of angiotensin II and activate endothelin, thus affecting vasomotor tone regulation[106,107]. Indeed, transgenic mice that express human chymase develop systemic hypertension, cardiac hypertrophy, and leucocytosis, confirming the important role of this enzyme in the regulation of blood pressure and inflammation[108]. The potentially destructive nature of the increased level of chymase is further proven by the observation that a high gene copy number in homozygotes is embryonically lethal[108]. Chymase can modulate inflammatory response by modification of cytokines and chemokines, e.g., IL-1β, IL-18, and CXCL7[109]. Moreover, it participates in ECM degradation and remodeling, either via direct cleavage of proteins such as fibronectin[110] or by activation of other enzymes, e.g., MMP-2 and MMP-9[111,112]. Investigation of the role of chymase in pulmonary hypertension pathogenesis confirmed the utility of chymase targeting as a promising therapeutic approach. In comparison to controls, inhibitor-treated animals showed improved hemodynamics and significantly reduced vascular remodeling. Decreased chymase activity resulted in lower levels of TGF-β1 and MMP-2, as well as diminished endothelin 1-mediated pulmonary vasoconstriction[113], providing hints into the mechanism behind the protective effect mediated by chymase inhibition.
OTHER SERINE PROTEASES
Serine proteases are a diverse family of proteolytic enzymes expressed not only in neutrophils and mast cells but also in other granulocytes, such as eosinophils and basophils. The role of eosinophils in PH is complex and context-dependent, as they have been implicated in both promoting[114] and preventing[115] the development of this condition. Animal models of pulmonary arterial hypertrophy have shown that depletion of eosinophils using anti-IL-5Rα antibodies can ameliorate vascular remodeling[116], and patients with eosinophilic COPD are at a 7-fold increased risk of PH compared to non-eosinophilic COPD[117]. These findings suggest a potential role for eosinophil-derived mediators in the pathogenesis of pulmonary hypertension. However, it is not yet clear whether eosinophil-derived serine proteases specifically contribute to this effect.
Basophil accumulation has also been observed in the lungs and pulmonary arteries of IPAH patients[26]. Basophils share many characteristics with mast cells, and it is possible that mast cell-targeted therapies exert their effects, at least in part, via modulation of basophil function. However, the specific role of basophils in pulmonary hypertension remains unclear. Most of the evidence to date supports the involvement of neutrophil-derived NSPs and MC-derived tryptase and chymase in the pathogenesis of PAH. However, it is important to consider that other serine proteases may also participate in the pathological processes underlying this disease, and further research is needed to fully elucidate their roles.
A summary of preclinical studies addressing anti-serine protease strategies in PH treatment can be found in Table 1.
Studies investigating therapeutic modulation of serine proteases in preclinical models of PH
| Target | Inhibition strategy | Preclinical PH model | Main outcomes |
| NE | Recombinant elafin | S100A4/Mts1 overexpression (develop PA neointimal lesion) -mice | Attenuation of the development of neointimal lesions[50] |
| Recombinant elafin | SuHx-rats | Improved hemodynamics and RV hypertrophy, regression of PA occlusive changes[68] | |
| Elafin overexpression | Chronic hox- mice | Decreased severity of pulmonary vascular disease[69] | |
| Inhibitors M249314 and ZD0892 | MCT-rats | Reversal of established PAH with normalization of PA pressure and structure[70] | |
| CtsG/PR3 | Elafin has also been shown to modulate the activity of PR3[75,76] The specific role of CtsG/PR3 in PAH has not been addressed | ||
| Chymase Tryptase | Ketotife (MC stabilizer) MC Knock Out | MCT-rats | Attenuation of PH and vascular remodeling[28] |
| Cromolyn (MC stabilizer) c-kit inhibitor PLX | MCT-rats | In preventive approach attenuated RVSP and decreased muscularization[29] | |
| FSLLRY-amide (PAR-2 antagonist blocks tryptase-PAR2 axis) | Chronic hox-mice | PAR-2 antagonist attenuated the development of PH and vascular remodeling[31] | |
| Cromolyn (MC stabilizer) TY-51469 (inhibitor) | MCT + aortocaval shunt -rats | Lower chymase activity was associated with less advanced pulmonary vascular remodeling and improved RV hemodynamics[93] | |
| BCEAB (inhibitor) | Bleomycin-induced PH due to lung fibrosis -hamsters | Reduced pulmonary vascular remodeling and lung fibrosis, lower expression of TGFβ1 and MMP2 in PAs[113] | |
| Chymostatin (inhibitor) | Cigarette smoke-induced COPD-PH-hamster | Reduction of chymase activity and Ang II concentration in the lung. Attenuated RVSP elevation and remodeling of pulmonary arterioles[118] | |
CONCLUDING
There is an urgent need for the identification and development of novel therapeutic strategies for PAH, as current treatments target primarily vasoconstriction rather than vasculopathy which underpins the pathology of this disease. Vasodilators ameliorate the symptoms, improve functional capacity and reduce hospitalizations, but do not protect patients from the development of progressive, occlusive vascular lesions and clinical deterioration[44,119]. Growing evidence supports the notion that dysregulated immunity and increased immune-cell-derived proteolytic activity play a central pathogenic role in the development and progression of vessel remodeling and PAH (summarized in Figure 1). Experimental models showed that inhibition of selected serine proteases, such as NE or chymase, can prevent, or in some instances even revert, vascular remodeling and disease progression[70,113]. Therefore, a strategy that modulates the effects of the aberrant activity of immune cell-derived proteases to reduce inflammation and preserve the structural integrity of the vessel wall holds great therapeutic promise. The development of anti-serine protease therapy is a rapidly advancing field of research. Despite significant efforts, designing an effective strategy has proven to be challenging. The multifaceted regulation of serine protease activity provides many targeting opportunities, but it also makes the design of an effective drug difficult. Furthermore, therapeutic targeting is complicated by the incredible complexity of the serine protease family and the fact that these enzymes are central regulators of many biological processes that are necessary for tissue homeostasis. This complexity requires a delicate balance between targeting serine proteases to achieve therapeutic benefits while avoiding disruption to normal tissue function. It is important to note that broad-spectrum inhibition of serine proteases can be detrimental, as demonstrated by Aprotinin, the administration of which significantly increases the risk of end-organ damage and death[120]. For safety reasons, a more refined, targeted approach needs to be favored. Elafin (NE and PR3 inhibitor) is currently being investigated in a phase 1 clinical trial for the treatment of PAH (ClinicalTrials.gov Identifier: NCT03522935). Cathepsin C (CtsC) makes another interesting target for modulating serine-proteases-mediated tissue damage and inflammation, as it is an upstream activator of NSPs, tryptase, chymase, and granzymes. Brensocatib, a CtsC inhibitor, has recently been confirmed as safe and effective in phase 2 clinical trials involving bronchiectasis patients[121]. Given the strong implication of neutrophil and MC serine proteases in the development of PAH, targeting CtsC to reduce proteolytic activity in the tissue - by preventing the conversion of zymogens to active enzymes[45] - emerges as a potential therapy to treat PAH. Genetic inactivation of CtsC in humans leads to Papillon-Lefèvre syndrome (PLS, OMIM: 245000), an autosomal recessive disorder in which NSP activity is minimal or absent[122]. Because the clinical consequences of this disorder are mild, small molecule inhibitors targeting CtsC activity are considered safe and represent an attractive therapeutic strategy to modulate NSP-mediated tissue damage and inflammation. However, given that neutrophil NSPs and MC serine proteases have a broad range of targets and affect various aspects of inflammation and tissue damage, inhibiting their activity could have immunomodulatory and anti-remodeling effects not only in PAH but also in many other diseases.
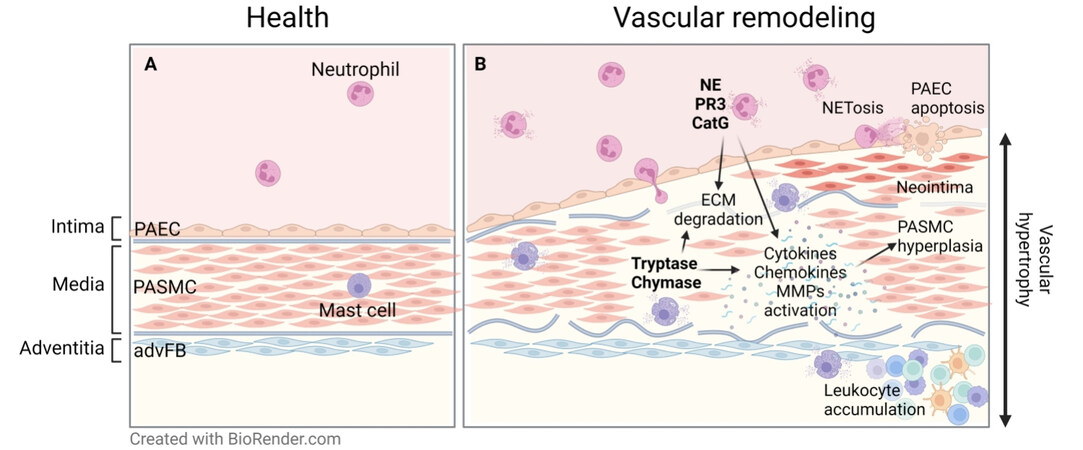
Figure 1. Involvement of proteases in pulmonary arterial remodeling in PH. (A) Schematic representation of a healthy arterial wall. (B) Arterial remodeling is accompanied by PASMC hyperplasia and ECM degradation and remodeling leading to the loss of vessel architecture. Immune cell-derived serine proteases can hydrolyze ECM proteins and modulate the bioavailability of cytokines, chemokines, and growth factors, thus regulating processes central to the development of vascular pathology. PAEC: Pulmonary arterial endothelial cells, PASMC: pulmonary arterial smooth muscle cells, advFB: adventitial fibroblasts, ECM: extracellular matrix, NE: neutrophil elastase, PR3: proteinase 3, CatG: cathepsin G. Graphical abstract has been created with BioRender.com.
DECLARATIONS
Authors’ contributionsWrote and edited the manuscript: Borek I
Provided a critical review of the manuscript: Kwapiszewska G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Authors 2023.
REFERENCES
1. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913.
2. Chang KY, Duval S, Badesch DB, et al. PHAR Investigators*. Mortality in pulmonary arterial hypertension in the modern era: early insights from the Pulmonary Hypertension Association Registry. J Am Heart Assoc 2022;11:e024969.
3. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011;8:443-55.
4. Hoffmann J, Wilhelm J, Marsh LM, et al. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension. Am J Respir Crit Care Med 2014;190:98-111.
5. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertensio. Cell Tissue Res 2017;367:643-9.
6. Hoffmann J, Marsh LM, Pieper M, et al. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am J Physiol Lung Cell Mol Physiol 2015;308:L1002-13.
7. Aiello VD, Gutierrez PS, Chaves MJ, Lopes AA, Higuchi ML, Ramires JA. Morphology of the internal elastic lamina in arteries from pulmonary hypertensive patients: a confocal laser microscopy study. Mod Pathol 2003;16:411-6.
8. Sutendra G, Dromparis P, Bonnet S, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl) 2011;89:771-83.
9. Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:261-72.
10. Perros F, Dorfmüller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;185:311-21.
11. Li C, Liu P, Song R, Zhang Y, Lei S, Wu S. Immune cells and autoantibodies in pulmonary arterial hypertension. Acta Biochim Biophys Sin (Shanghai) 2017;49:1047-57.
12. Becker MO, Kill A, Kutsche M, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med 2014;190:808-17.
13. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122:920-7.
14. Kimura H, Okada O, Tanabe N, et al. Plasma monocyte chemoattractant protein-1 and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2001;164:319-24.
15. Dorfmüller P, Zarka V, Durand-gasselin I, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2002;165:5534-9.
16. Balabanian K, Foussat A, Dorfmüller P, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med 2002;165:1419-25.
17. Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013;144:1346-56.
18. Tselios K, Gladman DD, Urowitz MB. Systemic lupus erythematosus and pulmonary arterial hypertension: links, risks, and management strategies. Open Access Rheumatol 2017;9:1-9.
19. Duong H, Bonham CA. Sarcoidosis-associated pulmonary hypertension: pathophysiology, diagnosis, and treatment. Clin Pulm Med 2018;25:52-60.
20. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J 2008;32:1371-85.
21. Tamosiuniene R, Tian W, Dhillon G, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 2011;109:867-79.
22. Daley E, Emson C, Guignabert C, et al. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med 2008;205:361-72.
23. Crnkovic S, Valzano F, Fließer E, et al. Single-cell transcriptomics reveals skewed cellular communication and phenotypic shift in pulmonary artery remodeling. JCI Insight 2022:7.
24. Kurakula K, Smolders VFED, Tura-Ceide O, Jukema JW, Quax PHA, Goumans MJ. Endothelial dysfunction in pulmonary hypertension: cause or consequence? Biomedicines 2021;9:57.
25. Le Hiress M, Tu L, Ricard N, et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. role of the macrophage migration inhibitory Factor/CD74 complex. Am J Respir Crit Care Med 2015;192:983-97.
26. Marsh LM, Jandl K, Grünig G, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2018;51:1701214.
27. Heath D, Trueman T, Sukonthamarn P. Pulmonary mast cells in mitral stenosis. Cardiovasc Res 1969;3:467-71.
28. Hoffmann J, Yin J, Kukucka M, et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 2011;37:1400-10.
29. Dahal BK, Kosanovic D, Kaulen C, et al. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir Res 2011;12:60.
30. Montani D, Perros F, Gambaryan N, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2011;184:116-23.
31. Kwapiszewska G, Markart P, Dahal BK, et al. PAR-2 inhibition reverses experimental pulmonary hypertension. Circ Res 2012;110:1179-91.
32. Ormiston ML, Chang C, Long LL, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012;126:1099-109.
33. Rätsep MT, Moore SD, Jafri S, et al. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am J Physiol Lung Cell Mol Physiol 2018;315:L977-90.
34. Qiu H, He Y, Ouyang F, Jiang P, Guo S, Guo Y. The role of regulatory T cells in pulmonary arterial hypertension. J Am Heart Assoc 2019;8:e014201.
35. Jandl K, Marsh LM, Mutgan AC, et al. Impairment of the NKT-STAT1-CXCL9 axis contributes to vessel fibrosis in pulmonary hypertension caused by lung fibrosis. Am J Respir Crit Care Med 2022;206:981-98.
36. Koudstaal T, van Hulst JAC, Das T, et al. DNGR1-Cre-mediated deletion of Tnfaip3/A20 in conventional dendritic cells induces pulmonary hypertension in mice. Am J Respir Cell Mol Biol 2020;63:665-80.
37. Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:897-908.
38. Ucero AC, Bakiri L, Roediger B, et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J Clin Invest 2019;129:3293-309.
39. Florentin J, Coppin E, Vasamsetti SB, et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol 2018;200:3612-25.
40. Özpelit E, Akdeniz B, Özpelit ME, et al. Prognostic value of neutrophil-to-lymphocyte ratio in pulmonary arterial hypertension. J Int Med Res 2015;43:661-71.
41. Harbaum L, Baaske KM, Simon M, et al. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm Med 2017;17:72.
42. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532-5.
44. Galiè N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J 2010;31:2080-6.
45. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726-59.
46. Pham CT. Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol 2008;40:1317-33.
47. Cowland JB, Borregaard N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol 1999;66:989-95.
48. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002;109:363-71.
49. Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 2000;12:201-10.
50. Kim YM, Haghighat L, Spiekerkoetter E, et al. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol 2011;179:1560-72.
51. Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation 2003;107:2829-36.
52. Rose F, Hattar K, Gakisch S, et al. Increased neutrophil mediator release in patients with pulmonary hypertension--suppression by inhaled iloprost. Thromb Haemost 2003;90:1141-9.
53. Sweatt AJ, Miyagawa K, Rhodes CJ, et al. Severe pulmonary arterial hypertension is characterized by increased neutrophil elastase and relative elafin deficiency. Chest 2021;160:1442-58.
54. Jandl K, Marsh LM, Hoffmann J, et al. Basement membrane remodeling controls endothelial function in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2020;63:104-17.
55. Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular matrix-bound basic fibroblast growth factor. J Cell Physiol 1996;166:495-505.
56. Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest 1980;66:859-62.
57. Zhu Y, Huang Y, Ji Q, et al. Interplay between Extracellular Matrix and Neutrophils in Diseases. J Immunol Res 2021;2021:8243378.
58. Geraghty P, Rogan MP, Greene CM, et al. Neutrophil elastase up-regulates cathepsin B and matrix metalloprotease-2 expression. J Immunol 2007;178:5871-8.
59. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 2006;6:541-50.
60. Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol 2009;183:2602-9.
61. Clancy DM, Sullivan GP, Moran HBT, et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep 2018;22:2937-50.
62. Fukuta T, Okada H, Takemura G, et al. Neutrophil elastase inhibition ameliorates endotoxin-induced myocardial injury accompanying degradation of cardiac capillary glycocalyx. Shock 2020;54:386-93.
63. Suzuki K, Okada H, Takemura G, et al. Neutrophil elastase damages the pulmonary endothelial glycocalyx in lipopolysaccharide-induced experimental endotoxemia. Am J Pathol 2019;189:1526-35.
64. Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 2012;18:1217-23.
65. Grechowa I, Horke S, Wallrath A, Vahl CF, Dorweiler B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J 2017;31:3868-81.
66. Ushakumari CJ, Zhou QL, Wang YH, et al. Neutrophil elastase increases vascular permeability and leukocyte transmigration in cultured endothelial cells and obese mice. Cells 2022;11:2288.
67. Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 2011;117:333-41.
68. Nickel NP, Spiekerkoetter E, Gu M, et al. Elafin reverses pulmonary hypertension via Caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med 2015;191:1273-86.
69. Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 2002;105:516-21.
70. Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med 2000;6:698-702.
71. Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett 1995;374:29-33.
72. Coeshott C, Ohnemus C, Pilyavskaya A, et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA 1999;96:6261-6.
73. Hurst LA, Dunmore BJ, Long L, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun 2017;8:14079.
74. Lane KB, Machado RD, Pauciulo MW, et al. International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000;26:81-4.
75. Wiedow O, Lüademann J, Utecht B. Elafin is a potent inhibitor of proteinase 3. Biochem Biophys Res Commun 1991;174:6-10.
76. Ying Q, Simon SR. Kinetics of the Inhibition of Proteinase 3 by Elafin. Am J Respir Cell Mol Biol 2001;24:83-9.
77. Wittamer V, Bondue B, Guillabert A, Vassart G, Parmentier M, Communi D. Neutrophil-mediated maturation of chemerin: a link between innate and adaptive immunity. J Immunol 2005;175:487-93.
78. Macvanin MT, Rizzo M, Radovanovic J, Sonmez A, Paneni F, Isenovic ER. Role of chemerin in cardiovascular diseases. Biomedicines 2022;10:2970.
79. Wittamer V, Franssen JD, Vulcano M, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med 2003;198:977-85.
80. Hanthazi A, Jespers P, Vegh G, et al. Chemerin added to endothelin-1 promotes rat pulmonary artery smooth muscle cell proliferation and migration. Front Physiol 2020;11:926.
81. Hanthazi A, Jespers P, Vegh G, et al. Chemerin influences endothelin- and serotonin-induced pulmonary artery vasoconstriction in rats. Life Sci 2019;231:116580.
82. Peng L, Chen Y, Li Y, et al. Chemerin regulates the proliferation and migration of pulmonary arterial smooth muscle cells via the ERK1/2 signaling pathway. Front Pharmacol 2022;13:767705.
83. Jankowich MD, Wu WC, Choudhary G. Association of elevated plasma endothelin-1 levels with pulmonary hypertension, mortality, and heart failure in african american individuals: The Jackson Heart Study. JAMA Cardiol 2016;1:461-9.
84. Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: novel cathepsin g-mediated catalytic activity that is resistant to inhibition. J Immunol 1998;160:1436-43.
85. de Man FS, Tu L, Handoko ML, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:780-9.
86. Bjornsson J, Edwards WD. Primary pulmonary hypertension: a histopathologic study of 80 cases. Mayo Clin Proc 1985;60:16-25.
87. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem 2000;275:6819-23.
88. Rigg RA, Healy LD, Chu TT, et al. Protease-activated receptor 4 (PAR4) activity promotes platelet granule release and platelet-leukocyte interactions. Platelets 2019;30:126-35.
89. Nufer O, Corbett M, Walz A. Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry 1999;38:636-42.
90. Richter R, Bistrian R, Escher S, et al. Quantum proteolytic activation of chemokine CCL15 by neutrophil granulocytes modulates mononuclear cell adhesiveness. J Immunol 2005;175:1599-608.
91. Stoeckle C, Sommandas V, Adamopoulou E, et al. Cathepsin G is differentially expressed in primary human antigen-presenting cells. Cell Immunol 2009;255:41-5.
92. Farha S, Sharp J, Asosingh K, et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012;2:220-8.
93. Bartelds B, van Loon RLE, Mohaupt S, et al. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012;141:651-60.
95. Schwartz LB, Irani AM, Roller K, Castells MC, Schechter NM. Quantitation of histamine, tryptase, and chymase in dispersed human T and TC mast cells. J Immunol 1987;138:2611-5.
96. Schwartz L, Lewis R, Austen K. Tryptase from human pulmonary mast cells. Purification and characterization. J Biol Chem 1981;256:11939-43.
97. Lützelschwab C, Pejler G, Aveskogh M, Hellman L. Secretory granule proteases in rat mast cells. Cloning of 10 different serine proteases and a carboxypeptidase A from various rat mast cell populations. J Exp Med 1997;185:13-29.
98. Pejler G, Rönnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood 2010;115:4981-90.
99. Kosanovic D, Dahal BK, Peters DM, et al. Histological characterization of mast cell chymase in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Pulm Circ 2014;4:128-36.
100. Compton SJ, Cairns JA, Holgate ST, Walls AF. Human mast cell tryptase stimulates the release of an IL-8-dependent neutrophil chemotactic activity from human umbilical vein endothelial cells (HUVEC). Clin Exp Immunol 2000;121:31-6.
101. Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1β and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol 1998;161:1939-46.
102. Iddamalgoda A, Le QT, Ito K, Tanaka K, Kojima H, Kido H. Mast cell tryptase and photoaging: possible involvement in the degradation of extra cellular matrix and basement membrane proteins. Arch Dermatol Res 2008;300 Suppl 1:S69-76.
103. Cairns JA, Walls AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest 1997;99:1313-21.
104. Abe M, Kurosawa M, Ishikawa O, Miyachi Y, Kido H. Mast cell tryptase stimulates both human dermal fibroblast proliferation and type I collagen production. Clin Exp Allergy 1998;28:1509-17.
105. Bagher M, Larsson-Callerfelt AK, Rosmark O, Hallgren O, Bjermer L, Westergren-Thorsson G. Mast cells and mast cell tryptase enhance migration of human lung fibroblasts through protease-activated receptor 2. Cell Commun Signal 2018;16:59.
106. D'Orléans-Juste P, Houde M, Rae GA, Bkaily G, Carrier E, Simard E. Endothelin-1 (1-31): from chymase-dependent synthesis to cardiovascular pathologies. Vascul Pharmacol 2008;49:51-62.
107. Simard E, Jin D, Takai S, Miyazaki M, Brochu I, D’Orle-Juste P. Chymase-dependent conversion of big Endothelin-1 in the mouse
108. Koga T, Urata H, Inoue Y, et al. Human chymase expression in a mice induces mild hypertension with left ventricular hypertrophy. Hypertens Res 2003;26:759-68.
109. Heutinck KM, ten Berge IJ, Hack CE, Hamann J, Rowshani AT. Serine proteases of the human immune system in health and disease. Mol Immunol 2010;47:1943-55.
110. Lazaar AL, Plotnick MI, Kucich U, et al. Mast cell chymase modifies cell-matrix interactions and inhibits mitogen-induced proliferation of human airway smooth muscle cells. J Immunol 2002;169:1014-20.
111. Frank BT, Rossall JC, Caughey GH, Fang KC. Mast cell tissue inhibitor of metalloproteinase-1 is cleaved and inactivated extracellularly by alpha-chymase. J Immunol 2001;166:2783-92.
112. Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem 2005;280:9291-6.
113. Kosanovic D, Luitel H, Dahal BK, et al. Chymase: a multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur Respir J 2015;46:1084-94.
114. Weng M, Baron DM, Bloch KD, Luster AD, Lee JJ, Medoff BD. Eosinophils are necessary for pulmonary arterial remodeling in a mouse model of eosinophilic inflammation-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2011;301:L927-36.
115. Shu T, Zhang J, Zhou Y, et al. Eosinophils protect against pulmonary hypertension through 14-HDHA and 17-HDHA. Eur Respir J 2023;61:2200582.
116. Ikutani M, Ogawa S, Yanagibashi T, et al. Elimination of eosinophils using anti-IL-5 receptor alpha antibodies effectively suppresses IL-33-mediated pulmonary arterial hypertrophy. Immunobiology 2018;223:486-92.
117. Alzghoul BN, As Sayaideh M, Moreno BF, et al. Pulmonary hypertension in eosinophilic versus noneosinophilic COPD. ERJ Open Res 2021;7:00772-2020.
118. Wang T, Han SX, Zhang SF, et al. Role of chymase in cigarette smoke-induced pulmonary artery remodeling and pulmonary hypertension in hamsters. Respir Res 2010;11:36.
119. Lan NSH, Massam BD, Kulkarni SS, Lang CC. Pulmonary arterial hypertension: pathophysiology and treatment. Diseases 2018;6:38.
120. Mangano DT, Tudor IC, Dietzel C. Multicenter Study of Perioperative Ischemia Research Group. The risk associated with aprotinin in cardiac surgery. N Engl J Med 2006;354:353-65.
121. Chalmers JD, Haworth CS, Metersky ML, et al. WILLOW Investigators. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med 2020;383:2127-37.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Borek I, Kwapiszewska G. Immune cell-derived serine protease as pathogenic drivers of vascular remodeling in pulmonary arterial hypertension. Rare Dis Orphan Drugs J 2023;2:5. http://dx.doi.org/10.20517/rdodj.2022.20
AMA Style
Borek I, Kwapiszewska G. Immune cell-derived serine protease as pathogenic drivers of vascular remodeling in pulmonary arterial hypertension. Rare Disease and Orphan Drugs Journal. 2023; 2(1): 5. http://dx.doi.org/10.20517/rdodj.2022.20
Chicago/Turabian Style
Borek, Izabela, Grazyna Kwapiszewska. 2023. "Immune cell-derived serine protease as pathogenic drivers of vascular remodeling in pulmonary arterial hypertension" Rare Disease and Orphan Drugs Journal. 2, no.1: 5. http://dx.doi.org/10.20517/rdodj.2022.20
ACS Style
Borek, I.; Kwapiszewska G. Immune cell-derived serine protease as pathogenic drivers of vascular remodeling in pulmonary arterial hypertension. Rare. Dis. Orphan. Drugs. J. 2023, 2, 5. http://dx.doi.org/10.20517/rdodj.2022.20
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 26
likes
Like This Article 26
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.