
Neutrophil serine proteases


Abstract
The identification and characterization of the four active neutrophil serine proteases (NSPs) have provided a better understanding of their roles in various physiological and pathological processes. The availability of appropriate tools such as substrates, inhibitors, and activity-based probes (ABPs) for studying their activity and functions in cells has become increasingly important. In this paper, the authors provide a comprehensive overview of the current knowledge on the tools available for studying NSPs. The substrates, inhibitors, and ABPs developed to date are described, including their strengths and limitations. The authors also discuss the potential implications of these tools for future research on NSPs, including their potential use in the development of new therapeutics for various diseases. Overall, this paper highlights the importance of understanding the activity and functions of NSPs and provides valuable information on the tools available for studying these proteases.
Keywords
INTRODUCTION
Neutrophils are the most abundant group of leukocytes normally occurring in human blood, which play an essential role in the innate branch of the immune system. The main functions of this type of cell are to fight infections and promote an inflammatory response to the onset of diseases caused by bacteria or fungi[1]. Neutrophils are activated by the emergence of specific danger signals such as inflammatory cytokines, molecules derived from pathogens, or a host tissue damage signal, and rapidly relocate from the peripheral blood into the inflammatory or damaged sites. As a part of the immune system’s first line of defense, neutrophils employ various mechanisms in order to eliminate the invading pathogens as well as to regulate inflammatory processes, including chemotaxis, phagocytosis, the release of reactive oxygen species (ROS) generated by the NADPH oxidase, formation of extracellular chromatin filaments containing granule-derived proteins (named neutrophil extracellular traps; NETs), and degranulation[1,2]. However, these protective mechanisms can also be destructive to host cells; therefore, neutrophil production, maturation, distribution, and disposal need to be strictly regulated[3].
Neutrophil granules contain numerous proteolytic enzymes of four different classes of proteases: cysteine proteases (e.g., cathepsin C), aspartyl proteases (e.g., cathepsin D, cathepsin E), metalloproteases
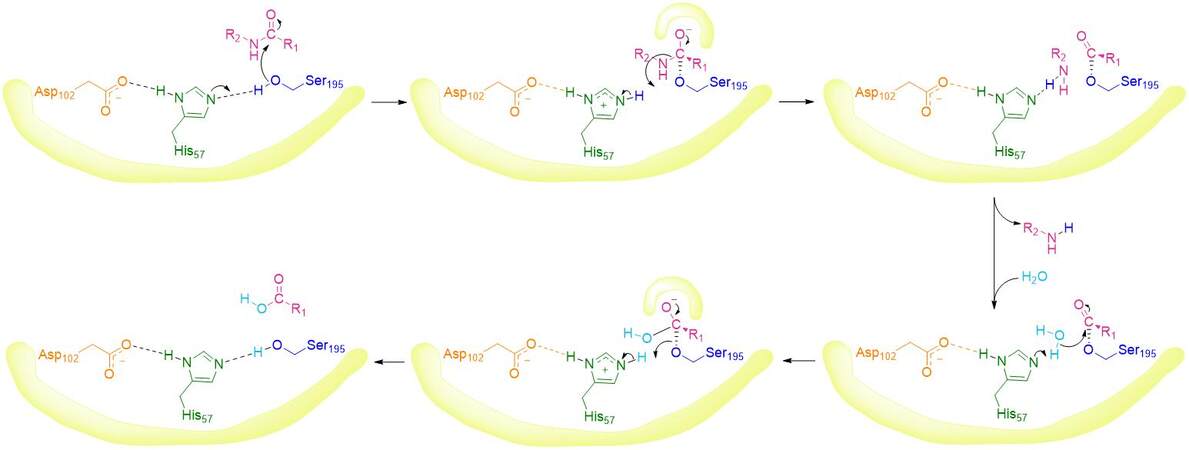
Figure 1. Mechanism of action neutrophil serine proteases.
Neutrophil serine proteases are synthesized as inactive zymogens in the bone marrow during early granulocyte development and contain a dipeptide structure at the N-terminal. These zymogens undergo maturation, which activates the enzymes and produces active neutrophil serine proteases. This N-terminal proteolytic processing of the inactive pre-form of NSPs takes place in the endoplasmic reticulum and depends on the activity of dipeptidyl peptidase I (DPPI). Dipeptidyl peptidase I, aminodipeptidase, also known as cathepsin C, is a cysteine protease located in the primary granules[5,9]. Under normal physiological conditions, the activity of NSPs must be controlled in order to avoid the development of pathological states or autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, chronic respiratory diseases (e.g., chronic obstructive pulmonary disease, emphysema, pulmonary fibrosis), and cancer[10-14].
HUMAN NEUTROPHIL ELASTASE
Human neutrophil elastase (HNE), also known as leukocyte elastase (EC 3.4.21.37), is a globular glycoprotein belonging to the chymotrypsin family. HNE is located in neutrophils, eosinophils, mast cells, monocytes, keratinocytes, and fibroblasts. This 29 kDa protease consists of a single polypeptide chain of 218 amino acids and two asparagine-linked carbohydrate chains localized at Asn95 and Asn144. The presence of four disulfide bridges and 19 arginine residues helps stabilize the structure, resulting in basic properties and an isoelectric point around 10-11 [Figure 2]. The primary structure of HNE exhibits homology with other NSPs, PR3 (57%) and CatG (37%). The substrate specificity of HNE is very similar to PR3, with both enzymes cleaving substrates after small aliphatic residues (Ala, Val, GABA, and norVal)[15].
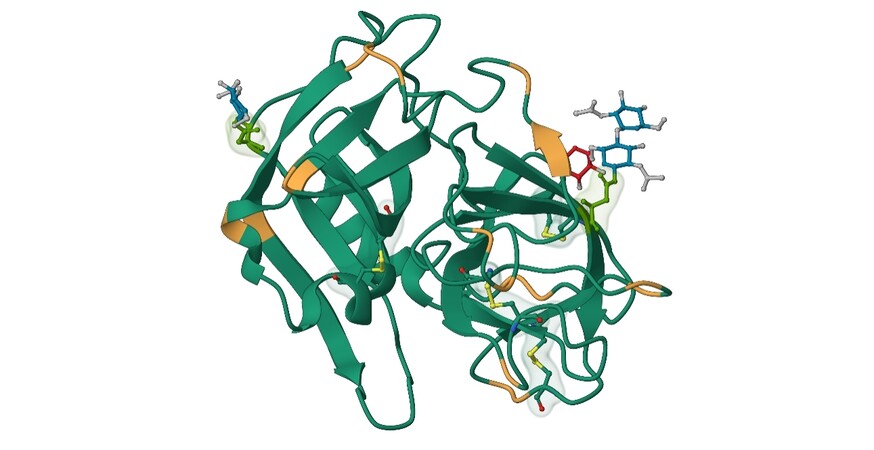
Figure 2. Structure of human neutrophil elastase; (green: Asn95 and Asn144, orange: Arg residues, yellow: disulfide bridges; based on 3Q76.pdb)[16].
This enzyme has a broad physiological function, including the degradation of elastin and other extracellular proteins like collagen (type I-IV), fibronectin, laminin and proteoglycans, and it has coagulation factors (fibrinogen, factors V, VII, XII, and XIII), plasminogen, immunoglobulins (IgG, IgA, and IgM), thrombomodulin, platelet, complement factors (C3, C5), complement receptors, and the coat protein of HIV (gp120)[15]. HNE is also responsible for the degradation or activation of many essential host immune molecules, such as interleukins (IL-1β, IL-2, IL-6, IL-8, IL-12p40, and IL-12p70) or tumor necrosis factor. Furthermore, HNE processes the surface of toll-like receptors TLR2, TLR4, CD14, and tumor necrosis factor receptors. It can also degrade other neutrophil proteases and proteases inhibitors, resulting in both their activation and inactivation[17].
HNE is an integral component of NETs, which form large web-like structures containing DNA, histones, and other granular proteins such as proteinase 3, myeloperoxidase, or high mobility group protein B1[18]. NETs play an essential role as a trap for extracellular pathogens in the first-line defense response of the innate immune system. There are forms and releases in a process known as NETosis, a unique type of cell death, after recognizing specific pathogens. NETs persistence is regulated by DNase I and DNase-like proteins, which are responsible for maintaining a balance between the formation and degradation of neutrophil extracellular traps[19,20]. Abnormal level of release NETs can be associated with the pathogenesis of different disorders, including lung injury[19,21], nephritis[22], cardiovascular disease[23], autoimmune disorders (e.g., rheumatoid arthritis, systemic lupus erythematosus, antiphospholipid syndrome[18,22]), as well as lung, breast or pancreatic cancer progression[24,25]. In the tumor microenvironment, the activity of release NE from NETs may also correlate with the formation of breast or colorectal cancer metastases[26,27]. Additionally, recent studies confirm that it is also related to COVID-19 due to SARS-CoV-2 infection can directly induce the increased formation of NETs in neutrophils[28-30].
The potent activity of NE is regulated by endogenous inhibitors, mainly by serpins such as α1-PI, α2-MG,
HNE Synthetic substrates
The most common substrates used for routine assay of HNE activity, which are hydrolytically stable at neutral pH, are chromogenic and fluorogenic peptide substrates. A synthesis of these simple compounds was initially described in 1979. The values of KM, kcat, and kcat/KM for the most potent chromogenic substrate MeO-Suc-Ala-Ala-Pro-Val-pNA (λex = 320 nm, λem = 490 nm) are respectively 0.14 mM, 17 s-1,
Wysocka et al. have described the synthesis and characterization of several fluorescent substrates displaying FRET for HNE[33]. The best parameters were obtained for PEG-FAM-Ala-Pro-Glu-Glu-Ile-Met--Asp-Arg-Gln-BAD [where PEG = 2-(2-(2-aminoethoxy)ethoxy)acetic acid, FAM = Lys(Fam)-OH, Fam = 5(6)-fluorescein, BAD = Ala(Bad)-OH], with the KM of 24.2 ± 3.1 µM, kcat of 6.2 ± 0.9 s-1, and kcat/KM of
In 2013 Sun et al. described a design and simple synthesis of a pentafluoroethyl conjugated 7-amino-4-trifluomethylcoumarin (AFC) as the first non-peptide-based fluorescent probe and a substrate for NE[34]. This low molecular weight compound, 2,2,3,3,3-pentafluoro-N-(2-oxo-4-(trifluoromethyl)-2H-chromen-7-yl) propenamide, with high specificity (KM = 20.45 ± 1.85 µM, kcat = 21.84 ± 4.37 min-1,
HNE ABPs and Inhibitors
Prolastin®, a purified α1-antitrypsin, is the first neutrophil elastase inhibitor available on the market used in the treatment of α1-AT deficiency[15]. Elaspol®, also known as sivelestat or ONO-5046 (1), is the second NE inhibitor approved for use and the first non-peptide inhibitor. Silvelestat is applied in Japan and South Korea for the treatment of ALI and ARDS associated with systemic inflammatory response syndrome. This molecule is known as a highly specific and effective competitive NE inhibitor with IC50 and Ki of 44 nM and 200 nM[35]. Alvelestat, AZD9668 (2), is a reversible oral highly selective NE inhibitor with IC50 and Ki of
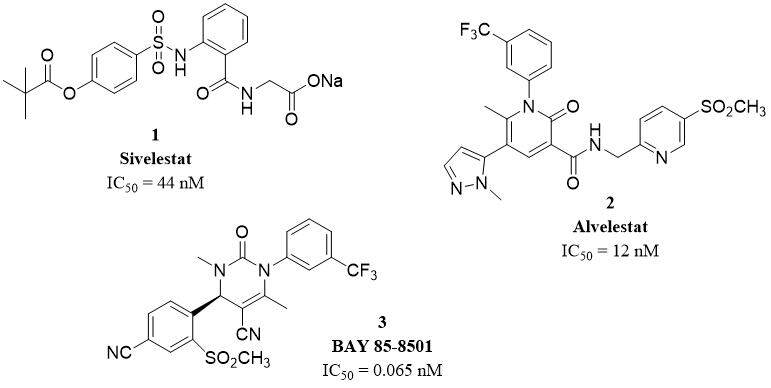
Figure 3. Structures of selected HNE inhibitors approved for use or in clinical trials[15].
Peptides chloromethyl ketone (CMK) are well-known inhibitors of HNE, with the most effective MeO-Suc-Ala-Ala-Pro-Val-CMK (4) with a kobs/[I] value of 922 M-1s-1[36] [Figure 4]. This group of inhibitors was useful for HNE structural studies and still serves as a cross-referent for newly developed HNE inhibitors. However, peptide-CMK has not been used in any clinical trials or therapy due to its high reactivity and potential toxicity[37].
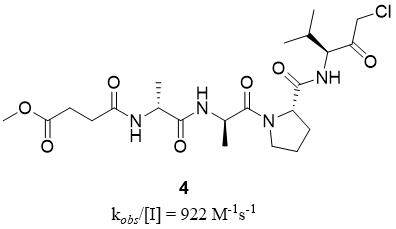
Figure 4. Structure of peptide chloromethyl ketone HNE inhibitor[37].
Compounds belonging to a group of diaryl esters of 1-aminoalkylphosphonate are well-known as highly selective and specific irreversible inhibitors of several serine proteases including NSPs. Compound 61 (5) is a peptide derivative of diaryl ester of 1-aminoalkylphosphonate [Figure 5], named 2-(4-(2-((S)-1-((S)-2-((R)-1-(Bis(4-(methylthio)phenoxy)-phosphoryl)-2-methylpropylcarbamoyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-ylamino)-2-oxoethoxy)phenoxy)acetic acid, and is an example of an irreversible and selective inhibitor of HNE with kinact/KI of 2,353,000 ± 89,000 M-1s-1. This group of inhibitors shows great stability in human plasma and PBS. The stability of irreversible interaction at elastase-inhibitor (compound 61) complex in time was confirmed; after 50 days of incubation, HNE recovered only 15% of initial activity[38].
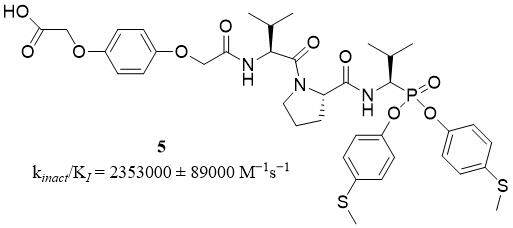
Figure 5. Structure of irreversible HNE inhibitor[38].
Diazaborines represent a new class of inhibitors of serine proteases with boron-based (B-N) heterocycle warheads, which can be used as a boronic acid replacement with similar activity and selectivity but better stability in plasma. This group has been described as reversible covalent inhibitors with selectivity toward HNE, and without activity against urokinase, trypsin, thrombin, kallikrein, and chymotrypsin. Cytotoxicity assay based on a human cell line HEK 293T confirms non-toxicity of diazaborines in concentrations of up to 100 µM after 48 h of incubation. The best results were obtained for the N-p-toluenesulfonyl substituent compound 18 (6) (IC50 = 2.7 ± 0.6 µM) and compound 19 (7) (IC50 = 0.7 ± 0.6 µM)[39] [Figure 6].
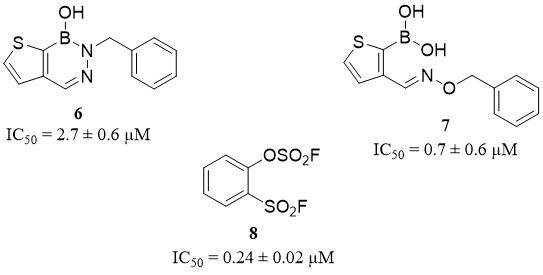
A promising class of covalent inhibitors of HNE with high selectivity is a group of sulfur fluoride exchange (SuFEx)able derivatives. From a set of 105 synthesized compounds, a simple benzenoid compound 24 (8),
Compounds belonging to benzenesulfonic acid derivatives are known as competitive inhibitors of HNE. According to Xu et al., from a series of synthesized ortho- and meta-substituted benzenesulfonic acids, only one, a compound 4f (9), shows moderate activity toward HNE, with an IC50 value of 35.2 µM[41] [Figure 7]. The cytotoxicity of the chosen compound was measured with five mammalian cancer cell lines, including MCF-7, BGC823, A549, HepG2, and HTC116, and no inherent cytotoxicity was observed[41].
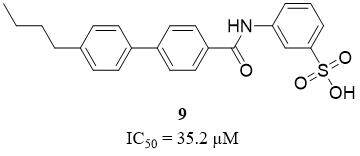
Figure 7. Structure of competitive HNE inhibitor[41].
Sulfonated nonsaccharide heparin mimetics are a group of novel potent, selective, non-competitive, and allosteric HNE inhibitors, without specificity toward other proteases, like plasmin, trypsin, and chymotrypsin, as well as heparin-binding coagulation proteins. Compound 3 (10), a hexa-sulfonated derivative, shows the most potent HNE inhibitor activity with an IC50 value of 0.22 ± 0.00 µM [Figure 8]. However, this compound does not scientifically affect the proliferation of the three human cell lines, including MCF-7, CaCo-2, and HEK-293 at a concentration of up to 10 µM[42].
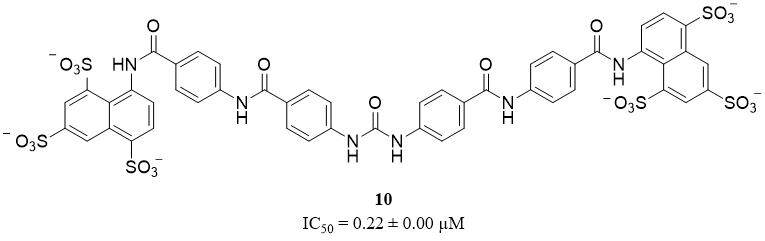
Figure 8. Structures of non-competitive allosteric HNE inhibitor[42].
In 2016, a small set of 1,2,3-triazole-based 4-oxo-β-lactam derivatives was synthesized and described as potential inhibitors and ABPs of HNE. The previous study confirms that the presence of oxo-β-lactam is essential for the interaction between the inhibitor and S1 pocket in HNE. The structure of the presented ABPs was based on the structure of the compound 2g (11), the most potent oxo-β-lactam HNE inhibitor with an IC50 of 14 ± 4 nM [Figure 9]. Finally, three different ABP structures were obtained - NBD-, fluorescein-, and biotin-tagged probes - respectively compound 3 (12) (IC50 = 56 ± 10 nM),
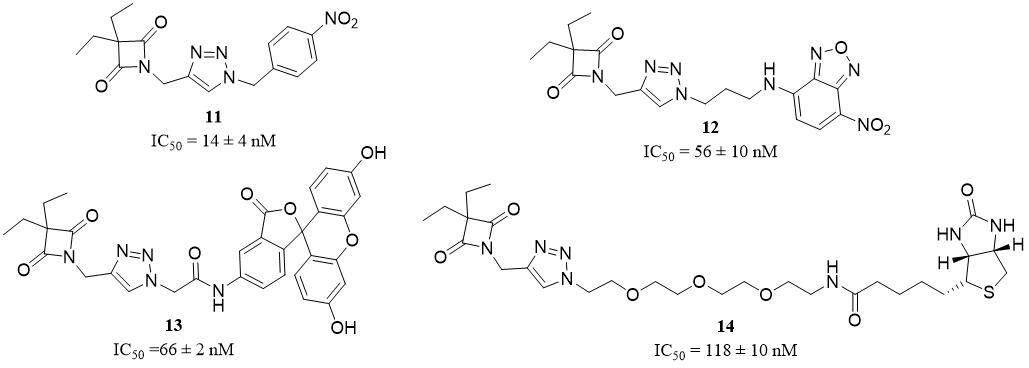
Figure 9. Structures of oxo-β-lactam HNE inhibitors[43].
In 2018, Schulz-Fincke et al. described a synthesis of compound 8 (15) as a potent inhibitor and ABP of HNE[44]. This compound belongs to sulfonyloxyphthalimide derivatives and contains coumarin 343 as a fluorescent tag [Figure 10]. The ability of inhibition was investigated with a spectroscopic assay with the chromogenic substrate MeOSuc-Ala-Ala-Pro-Val-pNA. This activity-base probe shows selective activity for HNE with a significant Ki value of 6.85 ± 0.39 nM and IC50 value of 0.0189 ± 0.0019 µM. The application of these probes enables the detection of NE in neutrophil cell lysate, with the proviso that its selectivity to PR3 is determined[44].
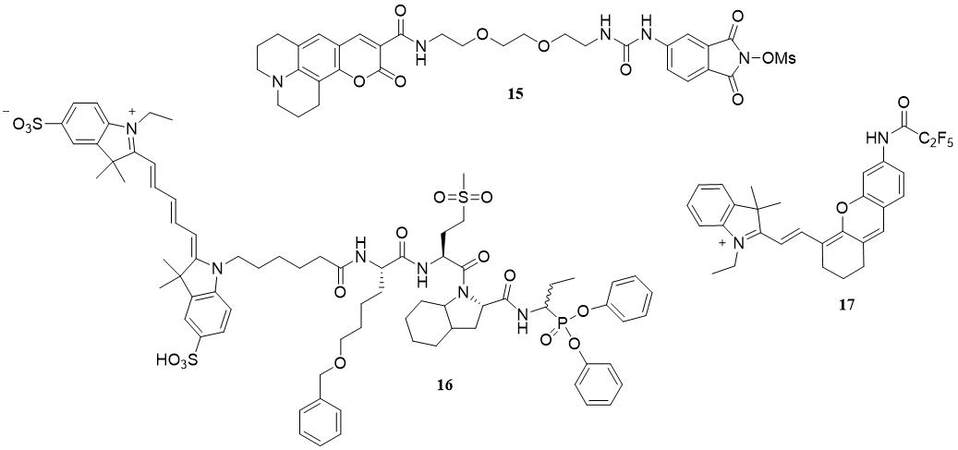
PK105b (16) is a fluorescent activity-based probe that contains sulfo Cy5 as a fluorophore, a region of unnatural specific amino acid, and diphenylphosponate as a warhead [Figure 10]. According to Anderson et al., this compound is an example of an irreversible HNE probe[45]. Application of PK105b facilitates the detection of NE activity in tissue lysates. Unfortunately, this APB was lack of selectivity to HNE, and cross-reactivity with other serine proteases is also observed[45].
Liu et al. posit that NEP (17) is a small-molecule-based near-infrared fluorogenic probe that could be used as a tool for highly specific and quick detection of HNE in vitro and in vivo[46] [Figure 10]. NEP is a molecule based on hemocyanin dye [Figure 10] with the optimum activity observed at pH 7.0 and a temperature of
Bacteria, plants, fungi or venomous animals are known as potential sources of new HNE inhibitors. Many of these compounds have been described in the literature; unfortunately, most of them have shown low potency of action, selectivity and stability in physiological conditions[47,48].
AvKTI is a peptide inhibitor of HNE, as well as trypsin, chymotrypsin, and plasmin, isolated from a spider Araneus ventricosus. This peptide with a sequence KDRCLLPKVTGPCKASLTRYYYDKDTKACVEFIYGGCRGNRNNFKQKDECEKACTDH is the first described Kunitz-type serine protease inhibitor with antifibrinolytic and anti-elastolytic activity. AvKTI shows the ability to inhibit HNE with an IC50 value of 446.93 nM and a Ki value of 169.07 nM. However, the inhibitory activity against plasmin was 44.4-fold stronger than against elastase (IC50 = 10.07 nM and
ShSPI, a natural peptide isolated from venomous Scolopendra hainanum is a typical Kazal-type protease inhibitor of HNE. This bioactive peptide is composed of 34 amino acids and contains a cysteine-stabilized
Loggerpeptins A-C and molassamide, natural peptides derived from marine cyanobacteria, consist of 19-membered ring cyclodepsipeptides containing the modified glutamic acid residue 3-amino-6-hydroxy-2-piperidone [Figure 11]. All peptides have been tested as inhibitors of HNE and compared with Sivelestat inhibition (IC50 = 0.06 µM). The kinetic test confirms the inhibitor activity of loggerpeptin A (18)
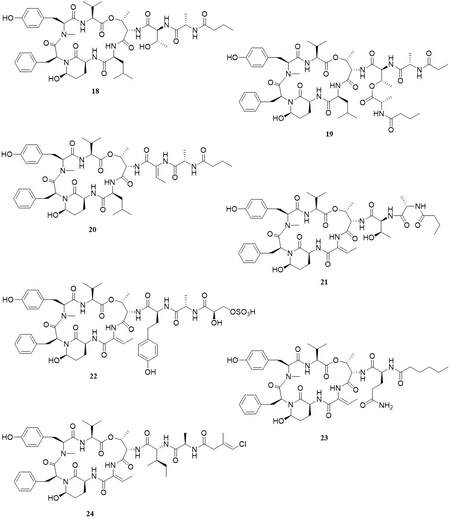
Figure 11. Structures of natural cyclodepsipeptide-type HNE inhibitors[48].
PROTEINASE 3
Proteinase 3 (PR3), also referred to as myeloblastin, azurophil granule protein-7, or p29b (EC 3.4.21.76), is the most abundant protease of the four NSPs mainly localized in azurophilic granules of neutrophils; however, it is also found in secretory vesicles. This protein is a 29 kDa chymotrypsin serine protease consisting of 222 amino acids and four disulfide bonds, which help stabilize the structure[54]. PR3 is the closest homolog of HNE; therefore, both enzymes have similar preferences at the S1 binding pocket for small aliphatic amino acid residues (Ala, Val, GABA, and norVal)[15].
One of the main functions of PR3 is the degradation of extracellular matrix components, including collagen, elastin, fibronectin, and laminin, leading to tissue remodeling and inflammation which is associated with chronic obstructive pulmonary disease (COPD) and granulomatosis with polyangiitis (GPA)[54,55]. Furthermore, PR3 is responsible for the cleavage of proinflammatory cytokines (IL-1β, IL-6, IL-8, IL-17,
Overexpression of PR3 can be associated with the development of autonomous cell growth in leukemia. PR3 also plays a major role as an autoantigen in many diseases, including granulomatosis with polyangiitis (GPA) and idiopathic interstitial pneumonia, and is also a target of anti-neutrophil cytoplasmic antibodies (ANCA) in vasculitis. In physiological conditions, the PR3 activity is regulated by natural inhibitors, including serpins (α1PI, monocyte neutrophil elastase inhibitor), chelonians (elafin, secretory leukocyte protease inhibitor), and α2-macroglobulin[54,55,57].
PR3 Synthetic substrates
For a long time, a synthesis of selective substrate for PR3 was difficult, mainly due to it being closely related to HNE and having a similar preference for small aliphatic amino acid residues at P1. In 2012, Epinette et al. published a new structure of selective FRET-type peptide substrate for PR3 without activity towards other serine proteases, especially HNE, ABZ-Val-Ala-Asp-norVal-Ala-Asp-Tyr-Gln-EDDnp with a KM value of 1.2 µM[58]. Another structure of selective FRET-type PR3 substrate consists of ABZ and Tyr(3-NO2) as a pair of donor and acceptor, ABZ-Tyr-Tyr-Abu-Asn-Glu-Pro-Tyr(3-NO2)-NH2 with a KM value of 3.2 mM and kcat/KM value of 1596 × 103 M-1s-1, was reported one year later[59]. A structure of simple chromogenic PR3 peptide substrates has also been developed, including Ac-Pro-Tyr-Asp-Ala-pNA and Ac-Asp-Tyr-Asp-Ala-pNA, with a kcat/KM value of 4201 ± 29.7 M-1s-1 and 162 ± 12.8 M-1s-1 respectively[60], as well as biotinylated derivative Bt-Val-Tyr-Asp-nVal-pNA with a kcat/KM value of 80510 ± 2973 M-1s-1[61].
PR3 ABPs and Inhibitors
Azapro-3 (25), ABZ-Val-Ala-Asp-aza(nor)Val-Ala-Asp-Tyr-Gln-Tyr(3-NO2), is an example of the first selective, non-covalent, reversible azapeptide inhibitor based on a structure of one of the FRET substrates for PR3 [Figure 12]. This competitive inhibitor shows high activity and selectivity for PR3 with a Ki value of 1.5 µM, with a nonsignificant inhibitory effect of HNE, and without any reactivity towards other proteases, including CatG, chymotrypsin, and granzyme B[58].
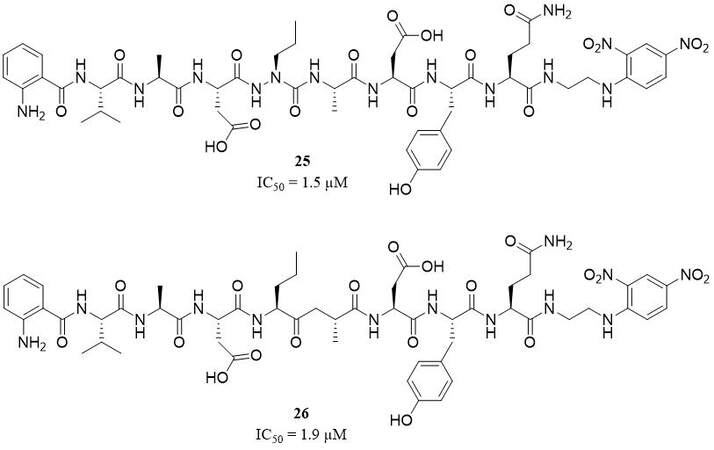
Another example of a PR3 inhibitor based on a sequence of its specific FRET substrate is ABZ-Val-Ala-Asp-(nor) Val[Ψ](COCH2)-Ala-Asp-Tyr-Gln-EDDnp (26), a ketomethylene peptide derivative [Figure 12]. This compound was characterized as a competitive and reversible inhibitor with an IC50 value of 1.91 µM, selective for PR3 over HNE[62].
Compound 6 (27), an analog of 2-aminobenzaldehyde oxime [Figure 13], was reported as the most potent non-peptide inhibitor for PR3 from a synthesized set of compounds, with an IC50 value of 0.22 ± 0.02 µM. However, this inhibitor showed a lack of selectivity due to better activity toward HNE
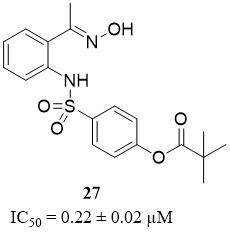
Figure 13. Structure of non-peptide PR3 inhibitor[63].
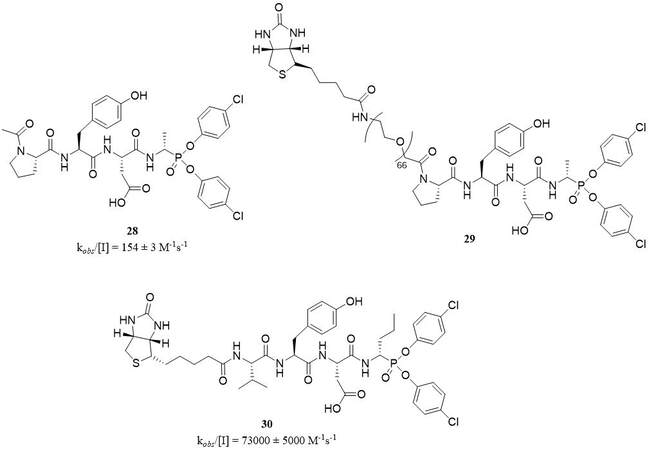
Structures based on aminophosphonates constitute a well-known group of potent and selective PR3 inhibitors, which have been recently reviewed[56]. The most important advantage of this class of inhibitors is their lack of reactivity with other proteases, including threonine, cysteine, aspartyl, and metalloproteinases. The first series of peptidyl derivatives α-aminoalkylphosphonate diaryl esters as specific, irreversible inhibitors of PR3 was synthesized in 2014. This most potent covalent inhibitor, which acts as the transition state analog, was Ac-Pro-Tyr-Asp-AlaP(O-C6H4-4-Cl)2 (28) with the kobs/[I] value of 154 ± 3 M-1s-1. A biotinylated derivative of this compound, Bt-[PEG]66-Pro-Tyr-Asp-AlaP(O-C6H4-4-Cl)2 (29), was an example of ABP, which can be used to visualize PR3 in native conditions, with kobs/[I] value of 1163 ± 0.1 M-1s-1
Several peptidyl biotinylated derivatives of α-aminoalkylphosphonate dsiaryl esters with a potential of ABPs for NSPs, including PR3 were described by Grzywa et al. Five of these structures,
Kinetic analysis of selected peptidyl biotinylated derivatives of α-aminoalkylphosphonate diaryl esters as inhibitors toward PR3, HNE, CatG
| Compound | PR3 kobs/[I] [M-1s-1] | HNE kobs/[I] [M-1s-1] | CatG kobs/[I] [M-1s-1] |
| Bt-Val-Pro-AbuP(O-C6H4-4-S-CH3)2 | 5.1 × 103 ± 350 | 4.2 × 105 ± 7500 | 5%[a] |
| Bt-Val-Pro-ValP(O-C6H5)2 | 1.6 × 103 ± 100 | 1.8 × 105 ± 7850 | N.I. |
| Bt-Val-Pro-ValP(O-C6H4-4-S-CH3)2 | 1.6 × 104 ± 1200 | 5.5 × 105 ± 25000 | N.I. |
| Bt-Val-Pro-LeuP(O-C6H4-4-COOCH3)2 | 1.9 × 104 ± 2300 | 2.1 × 105 ± 750 | 1.6 × 103 ± 125 |
| Bt-LC-Suc-Phe-Val-Thr-(4Gu)PhgP(O-C6H4-4-S-CH3)2 | 2.1 × 103 ± 100 | N.I. | 2.4 × 102 ± 25 |
In 2018, the same research group reported a structure of a new biotinylated PR3 inhibitor, Bt-Val-Tyr-Asp-nValP(O-C6H4-4-Cl)2 (30), with kobs/[I] value of 73,258 ± 5,342 M-1s-1, and without significant inhibitory activity toward HNE[61] [Figure 14].
The most potent phosphonic inhibitor and fluorescent ABP against PR3 were designed by Kasperkiewicz et al.[65]. The structure of compound PK302 (31), with a kobs/[I] value of 1.4 × 106 ± 11 M-1s-1, contains an optimal substrate sequence consisting of natural and unnatural amino acids [Figure 15]. However, this probe was not selective, and it showed weak reactivity toward HNE (kobs/[I] = 300 ± 50 M-1s-1)[65].
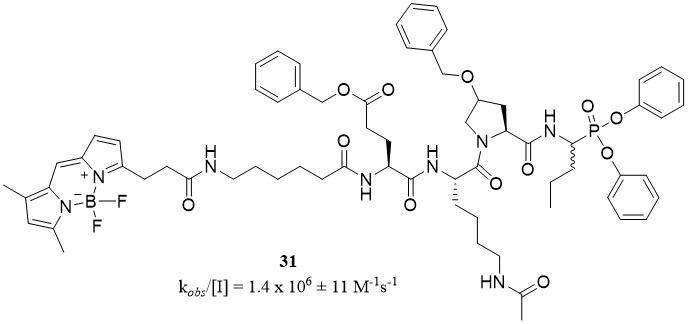
Figure 15. Structures of fluorescent PR3 activity-based probe[65].
Cyclic peptide compounds containing SFTI-variants can be an attractive group of new reversible, competitive inhibitors of PR3 with high stability in human serum. The most potent selective inhibitor toward PR3, from a synthesized set, was compound 3, c[GTCTAbuSIPPICNPN], a cyclized Gly1-Asn14 including a disulfide bond between Cys3-Cys11, with a Ki value of 9.8 ± 1.2 nM[66].
Pro3-SBP (32) is a near-infrared fluorescent (NIRF) substrate-based probe (SBP) designed and synthesized as a tool for monitoring active, secreted human PR3. The structure of a peptide hairpin loop consists of a specificity region (a PR3 recognition sequence) and an electrostatic zipper, driving a close vicinity of the FRET couple (sulfoCy5.5 and QSY21) [Figure 16]. Pro3-SBP (λex = 684 nm, λem = 710 nm) was specifically hydrolyzed by extracellular PR3 with kcat/KM value of 440,000 ± 5,500 M-1s-1. Unfortunately, hydrolysis by HNE was also observed in the minority (kcat/KM = 33,000 ± 3,000 M-1s-1)[67].
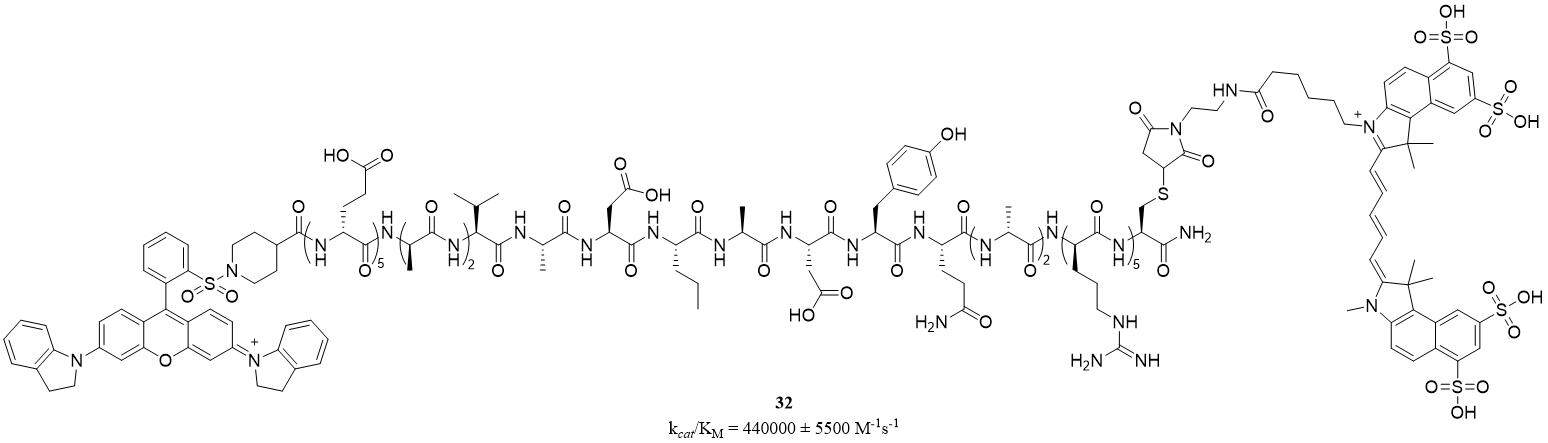
Figure 16. A structure of near-infrared fluorescent substrate-based probe of PR3[67].
CATHEPSIN G
Cathepsin G is expressed in neutrophils, mastocytes and monocytes. It is synthesized as a zymogen and is activated during packaging into granules by dipeptidyl peptidase I (DPPI) to an active form involved in several important physiological processes within the human body. CatG active site is typical of serine proteases and is composed of serine, histidine and aspartic acid. CatG structure represents the chymotrypsin fold[68]. CatG - similar to other neutrophil serine proteases - plays an important role in host defense mechanisms. CatG also mediates -platelet aggregation[69].
CatG activity also includes proteolytic processing of Ang-I and angiotensinogen and the production of angiotensin-II (Ang-II)[70]. CatG is also responsible for the activation of matrix metalloproteinases
As CatG plays an important role in many biological events, it has become a target for the development of a set of peptides and peptidomimetics substrates, inhibitors and ABPs.
CatG Synthetic substrates
The first studies on CatG and inhibitors revealed unusual acceptance of both the bulky hydrophobic side chains of Phe or Leu, in the S1 pocket, and the basic side chains of Lys and Arg[74,75]. Solving of the crystal structure of the complex between human cathepsin G and peptidyl phosphonate inhibitor Suc-Val-Pro-PheP-(OPh)2 showed that this dual specificity is caused by Glu226 residue, which is located at the bottom of the S1 pocket. This Glu226 residue interacts either with the basic side chain of Lys/Arg or with an edge of the aromatic ring of Phe[76].
In 1998, Polanowska et al. investigated the dual trypsin- and chymotrypsin-like specificities of CatG[74]. They synthesized a library of tetrapeptide p-nitroanilide substrates (Suc-Ala-Ala-Pro-Aaa-p-nitroanilide) to examine the specificity of the S1 binding pocket of human cathepsin G[74]. Their studies have shown/showed that cathepsin G can recognize both large hydrophobic and basic side chains, with a slight preference for Lys over Phe (1.3-fold) and Arg over Leu (1.1-fold). The best substrate from their studies was Suc-Ala-Ala-Pro-Lys-pNA (33) (KM = 2.75 × 10-3 M and kcat/KM 1,483 M-1s-1).
In 2007, Wysocka et al. presented studies about new chromogenic substrates of CatG[75]. The combinatorial chemistry methods enable the production of new, sensitive cathepsin G substrates. The introduction of the non-proteinogenic amino acid residue (4-guanidine-L-phenylalanine) in position P1 increases activity twice as high as in the case of Phe (Ac-Phe-Val-Thr-Gnf-Anb-NH2 (34) KM = 203 µM and kcat/KM 95,300 M-1s-1; Ac-Phe-Val-Thr-Phe-Anb-NH2 KM = 464 µM and kcat/KM 7,900 M-1s-1). The additionally obtained substrate is not cleaved by proteinase 3, human leukocyte elastase or chymotrypsin. A further modification by replacing the acetyl moiety with a residue of 7-methoxycoumarin-4-yl acetic acid (Mca) that served as a fluorescence donor and substrate elongation led to the Mca-Phe-Val-Thr-Gnf-Ser-Trp-ANB-NH2 (35), the sequence with a specificity constant (KM = 2 µM, kcat/KM = 252 × 103 M-1s-1) which is two orders of magnitude higher than that of the parent compound. The detection limit of CatG using this substrate is 70 pM[76]. Later in 2019, Groborz et al. developed new fluorogenic substrates of CatG based on a fluorescence quenching mechanism[77]. The most active inhibitor from their studies was OS-CG_11 (36) with kcat/KM = 206,854 M-1s-1 [Figure 17]. This substrate was also active against PR3 and displayed kcat/KM 146,824 M-1s-1[77].
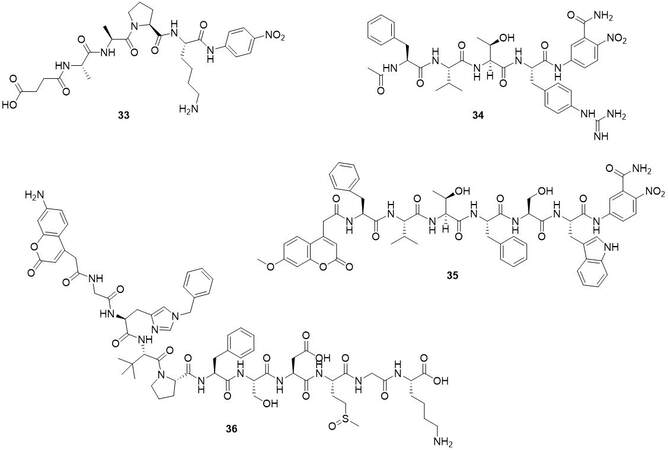
Figure 17. Synthetic substrates of CatG.
CatG ABPs and Inhibitors
In 1991, two phosphonic inhibitors of CatG were reported[78]. The analog of phenylalanine: Cbz-PheP(OPh)2 (37) and phenylglycine: Cbz-PhgP(OPh)2 (38) displays similar poor activity against CatG (kobs/I = 76 M-1s-1 and kobs/I = 91 M-1s-1 respectively) [Figure 18]. Further incorporation of the basic functional group into the aromatic side chain together with phenyl esters modification and peptide chain elongation resulted in the inhibitor Ac-Phe-Val-Thr-(4-guanidine)PhgP(OC6H4-4-S-Me)2 (39) with kobs/I = 256,000 M-1s-1[79] [Figure 18]. Based on this inhibitor, Grzywa et al. developed a low-molecular-weight activity-based probe: Bt-LC-Suc-Phe-Val-Thr-(4Gu)PhgP(O-C6H4-4-S-CH3)2 (40) [Table 2] with kobs/I = 240 M-1s-1 which displayed absolute specificity toward CatG in western blotting analysis among two other neutrophil proteases- HNE and PR3[64] [Figure 18]. Another phosphonate base ABPs was reported by the Drag group in 2017[65]. From a series of compounds with a different fluorophore inhibitor 202 (41), it showed the best potency with
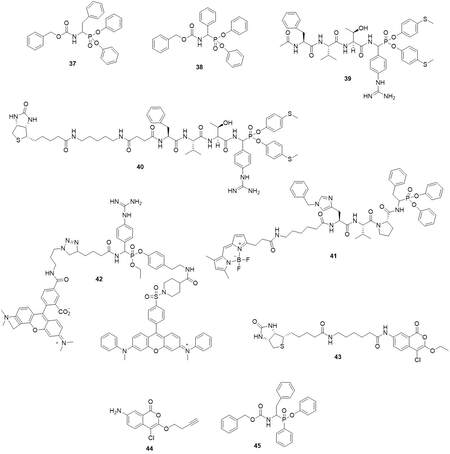
Figure 18. Inhibitors and ABPs of CatG.
Characterization of human neutrophil elastase inhibitors approved for use and in clinical trials
| Name | Therapeutic application | Development phase | Features |
| Prolastin® (α1-antitrypsin)[15] | α1-antitrypsin deficiency and clinical evidence of emphysema | FDA approved | peptide reversible NE inhibitor recommended dose: 60 mg/kg body weight once a week (for Prolastin-C Liquid) |
| Elaspol® (Sivelestat/ ONO-5046)[35] | acute lung injury and acute respiratory distress syndrome associated with the systemic inflammatory response syndrome | approved for use in Japan and South Korea | non-peptide competitive NE inhibitor IC50 44 nM; Ki 200 nM |
| Alvelestat (AZD9668)[15] | bronchiolitis obliterans syndrome | II phase clinical trials | reversible oral NE inhibitor IC50 12 nM; Ki 9.4 nM |
| BAY 85-8501[15] | non-cystic fibrosis bronchiectasis | II phase clinical trials | reversible NE inhibitor, IC50 0.065 nM; Ki 0.08 nM |
In 2015, Serim et al. reported quenched fluorescent activity-based probes based on mixed alkyl-aryl phosphonate esters[80] [Figure 18]. Compound 15 (42) could label the CatG in SDS-PAGE gel.
Isocoumarins equipped with detection tags were reported as CatG ABPs. Compound BIC5 showed
Recently, Kahler et al. developed aminomethyl phosphinate esters as inhibitors of serine protease. This group of compounds can interact with prime enzyme site. Primary compound testing revealed that inhibitor 13a (45) can react with CatG[83] [Figure 18].
NSP4
Human NSP4 (encoded by the gene PRSS57) is a trypsin-fold protease stored in neutrophil azurophilic granules[84,85]. Since the NSP4 discovery in 2012 by the D.E.Jenne research group, numerous studies regarding NSP4 biology, substrate specificity and inhibitors development have been done. However, still, blank spots remain to be filled. NSP4, unlike other NSPs, has been conserved for over 400 million years from bony fish to humans[86]. NSP4 is the only known enzyme that cleaves substrates with post-translationally modified arginine residues, such as methylarginine and citrulline[87]. These attributes suggest that NSP4 could have another potential function, not typical of NSPs. In 2020, AhYoung et al. demonstrated that NSP4 plays an essential role in mast cell biology[88]. Their studies have shown/showed that NSP4 is present during early mast cell development and is critical for the regulation of levels of histamine and serotonin in the secretory granules of the developing mast cells. They discovered that NSP4 deficiency causes protection against mast cell/histamine-dependent vascular leakage. These findings open new opportunities for therapeutic intervention of mast cell-dependent allergic and autoimmune diseases.
Synthetic substrates
Jenne’s pioneering work revealed that NSP4 can hydrolyze synthetic substrates: Boc- Ala-Pro-Nva-thiobenzyl ester -typical substrate for HNE and PR3[84]. Surprisingly, NSP4 showed/shows strong arginine preference at the P1 position. Tyr-Arg-Phe- Arg-AMC was efficiently hydrolyzed by NSP4. The replacement of arginine residue with lysine results in an almost complete lack of enzyme activity. Further studies explained the unique dual cleavage specificity of NSP4. In 2014, Lin et al. revealed an unusual mechanism of substrate recognition[87]. The arginine recognition by NSP4 relies on two important structural features[87]. The occluded S1 pocket and the H-bond acceptors around the P1-arginine guanidinium group. The substrate P1-arginine tits into the S1 pockets thanks to adopting a noncanonical ‘‘up’’ conformation, which is stabilized by a solvent-exposed H-bond network. Interestingly, except for NSP4, all other trypsin-like proteases rely heavily on the salt bridge within a/the well-defined S1 pocket. As a result of the unusual substrate recognition mechanism, NSP4 can hydrolase substrates with methylarginine or citrulline at the P1 position. This suggests one of the possible roles played by NSP4 protease, which is processing the post-translationally modified proteins. The substrate used during the studies was MCA-PEG-Ile-Arg-Arg-Ser-Ser-Tyr-Ser-Phe-Lys (Dnp)-Lys (KM = 13.8 µM and kcat/KM 10,000 M-1s-1).
The first studies to deeply investigate the substrate preferences of NSP4 were made by Kasperkiewicz et al.[89]. They synthesized the library of fluorogenic substrates with a fixed arginine residue at the P1 position. Activity studies showed that at the P2 position, substrates with proline residue were the most active among proteinogenics aminoacids. The substrates with the proline analog -Oic were the most active. P3 position revealed broad tolerance where Val, Ile, Tyr, Arg, Lys and Phe were the most active. The best non- proteinogenic amino acid was the phenylalanine derivative with a guanidine group in a/the para position (Phe(guan)). Homocyclohexylalanine (hCha) was found as the optimal amino acid at the P4 position. The most active substrate from the studies was Ac-hCha-Phe(guan)-Oic-Arg-ACC
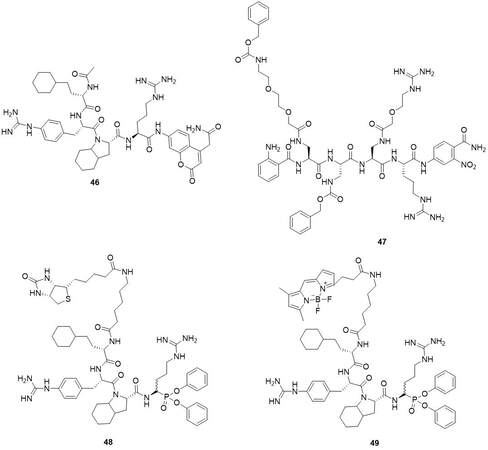
Figure 19. Synthetic substrates and ABPs of NSP4.
One year later, Wysocka et al. presented a new library of the PEGylated substrates of NSP4 protease[90]. From the library of a novel type of peptidomimetics composed of diaminopropionic acid residues modified with structurally diverse heterobifunctional polyethylene glycol chains (DAPEG), substrate 3 (47) was the most active with (Km = 7.6 µM and kcat/KM 13,1579 M-1s-1) [Figure 19].
Inhibitors and ABPs
Jenne et al. found that among natural inhibitors of NSPs - NSP4 was most efficiently inhibited by antithrombin-heparin. α1-proteinase inhibitor (α1-antitrypsin) and C1 inhibitor can block enzymatic activity of NSP4[84].
The first synthetic inhibitor and ABP were reported by Kasperkiewicz et al.[89]. The substrate conversion into the diphenyl phosphonate probe with a biotin tag led to the Biot-Ahx-hCha-Phe(guan)-Oic-ArgP(OPh)2 (48) (kobs/I = 3.8 × 106 M-1s-1 against NSP4 and kobs/I = 3.0 × 103 M-1s-1 against CatG and inactive against PR3 an HNE)[89] [Figure 19].
Further studies by Kasperkiewicz et al. about the development of the fluorescent ABPs of NSP lead to the compounds which are related to 401 and equipped with different N-Terminal fluorescent tags[65]. ABP with BODIPYFL fluorophore (49) was the most active with
SUMMARY
NSPs are protein-degrading enzymes but also take part in a wide variety of pathophysiological processes. Thus, their inhibitors are considered potential therapeutics. Despite the fact of extensive research on substrates, inhibitors and ABPs of NSPs, there is no FDA-approved drug acting as an NSPs inhibitor.
The most advanced research was done in the field of HNE inhibitors, where inhibitors of this enzyme entered clinical trials. However, clinical trials of HNE inhibitors have faced challenges and failures. For example, a phase II clinical trial of an HNE inhibitor called AZD9668 (Alvelestat) in patients with COPD failed to show significant improvement in lung function or other clinical endpoints compared to a placebo. Another phase II clinical trial of an HNE inhibitor called ONO-6818 in patients with cystic fibrosis did not meet its primary endpoint of improving lung function compared to a placebo. A phase I clinical trial of an HNE inhibitor called M3364 in patients with advanced solid tumors showed that the drug was well-tolerated and had some anti-tumor activity, with one patient experiencing a partial response and several others having stable disease.
The reasons for the failure of HNE inhibitors in clinical trials are multifactorial. One possible explanation is the complexity and heterogeneity of the diseases that HNE inhibitors are targeting. For example, COPD and cystic fibrosis are characterized by chronic inflammation, but there are multiple underlying causes and factors that contribute to the disease pathogenesis. Therefore, targeting HNE alone may not be sufficient to achieve clinical benefits. Another challenge is the difficulty in achieving optimal pharmacokinetic and pharmacodynamic properties of HNE inhibitors. In some cases, the HNE inhibitors may not be sufficiently potent or selective to effectively inhibit HNE in vivo, or they may be rapidly metabolized or eliminated from the body.
The latest studies have indicated the possibility of using elastase inhibitors in the treatment of acute respiratory distress syndrome (ARDS), which is a potentially life-threatening complication of respiratory infections such as COVID-19. ARDS is characterized by severe inflammation in the lungs, which can cause respiratory failure and other serious complications. Researchers have been investigating various ways to prevent or treat ARDS in COVID-19 patients, and one promising avenue of research has focused on inhibiting elastase activity. Several studies have explored the potential of elastase inhibitors as a therapeutic strategy for COVID-19-associated ARDS; however, there is no approved clinical treatment of ARDS with elastase inhibitors.
Further studies on NSPs (neutrophil serine proteases) are crucial for better understanding their role in various pathological conditions and for the development of new NSP-targeted drugs. Neutrophil serine proteases are enzymes released by immune cells, such as neutrophils, in response to infection or inflammation. They play an important role in host defense by breaking down and destroying invading pathogens. However, the excessive or uncontrolled activity of NSPs can lead to tissue damage, inflammation, and other pathological conditions. Therefore, the development of NSP-targeted drugs has emerged as a promising therapeutic strategy for various diseases, including chronic obstructive pulmonary disease (COPD), cystic fibrosis, and inflammatory bowel disease. NSP-targeted drugs can potentially limit the harmful effects of NSPs on host tissues while preserving their beneficial antimicrobial properties.
DECLARATIONS
Authors’ contributionsConception and writing of the article: Torzyk K, Skoreński M
Manuscript preparation, final correction: Sieńczyk M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by Statut Funding No. 8211104160.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. Gómez-Moreno D, Adrover JM, Hidalgo A. Neutrophils as effectors of vascular inflammation. Eur J Clin Invest 2018;48 Suppl 2:e12940.
3. Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L. Update on neutrophil function in severe inflammation. Front Immunol 2018;9:2171.
4. Majewski P, Majchrzak-Gorecka M, Grygier B, Skrzeczynska-Moncznik J, Osiecka O, Cichy J. Inhibitors of serine proteases in regulating the production and function of neutrophil extracellular traps. Front Immunol 2016;7:261.
6. Majchrzak-Gorecka M, Majewski P, Grygier B, Murzyn K, Cichy J. Secretory leukocyte protease inhibitor (SLPI), a multifunctional protein in the host defense response. Cytokine Growth Factor Rev 2016;28:79-93.
7. Liu R, Chen L, Wu W, Chen H, Zhang S. Neutrophil serine proteases and their endogenous inhibitors in coronary artery ectasia patients. Anatol J Cardiol 2016;16:23-8.
8. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-Driven inflammation. Cell Rep 2019;27:3646-3656.e5.
9. Benarafa C, Simon HU. Role of granule proteases in the life and death of neutrophils. Biochem Biophys Res Commun 2017;482:473-81.
10. Gao S, Zhu H, Zuo X, Luo H. Cathepsin G and Its role in inflammation and autoimmune diseases. Arch Rheumatol 2018;33:498-504.
11. Twaddell SH, Baines KJ, Grainge C, Gibson PG. The emerging role of neutrophil extracellular traps in respiratory disease. Chest 2019;156:774-82.
12. Gramegna A, Amati F, Terranova L, et al. Neutrophil elastase in bronchiectasis. Respir Res 2017;18:211.
13. Lerman I, Hammes SR. Neutrophil elastase in the tumor microenvironment. Steroids 2018;133:96-101.
14. Taylor S, Dirir O, Zamanian RT, Rabinovitch M, Thompson AAR. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front Med (Lausanne) 2018;5:217.
15. Crocetti L, Quinn MT, Schepetkin IA, Giovannoni MP. A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014-2018) and their therapeutic applications. Expert Opin Ther Pat 2019;29:555-78.
16. Hansen G, Gielen-Haertwig H, Reinemer P, Schomburg D, Harrenga A, Niefind K. Unexpected active-site flexibility in the structure of human neutrophil elastase in complex with a new dihydropyrimidone inhibitor. J Mol Biol 2011;409:681-91.
17. Domon H, Terao Y. The role of neutrophils and neutrophil elastase in pneumococcal pneumonia. Front Cell Infect Microbiol 2021;11:615959.
18. Wang W, Su J, Yan M, Pan J, Zhang X. Neutrophil extracellular traps in autoimmune diseases: Analysis of the knowledge map. Front Immunol 2023;14:1095421.
19. Scozzi D, Liao F, Krupnick AS, Kreisel D, Gelman AE. The role of neutrophil extracellular traps in acute lung injury. Front Immunol 2022;13:953195.
20. Zonta YR, Dezen ALO, Della Coletta AM, et al. Paracoccidioides brasiliensis releases a DNase-Like protein that degrades NETs and allows for fungal escape. Front Cell Infect Microbiol 2020;10:592022.
21. Yan S, Li M, Liu B, Ma Z, Yang Q. Neutrophil extracellular traps and pulmonary fibrosis: an update. J Inflamm (Lond) 2023;20:2.
22. Moore S, Juo HH, Nielsen CT, Tyden H, Bengtsson AA, Lood C. Role of neutrophil extracellular traps regarding patients at risk of increased disease activity and cardiovascular comorbidity in systemic lupus erythematosus. J Rheumatol 2020;47:1652-60.
23. Bonaventura A, Montecucco F, Dallegri F, et al. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc Res 2019;115:1266-85.
24. Quail DF, Amulic B, Aziz M, et al. Neutrophil phenotypes and functions in cancer: a consensus statement. J Exp Med 2022:219.
25. Cedervall J, Herre M, Dragomir A, et al. Neutrophil extracellular traps promote cancer-associated inflammation and myocardial stress. Oncoimmunology 2022;11:2049487.
26. Yang C, Wang Z, Li L, et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J Immunother Cancer 2021;9:e002875.
27. Okamoto M, Mizuno R, Kawada K, et al. Neutrophil extracellular traps promote metastases of colorectal cancers through activation of ERK signaling by releasing neutrophil elastase. Int J Mol Sci 2023;24:1118.
28. Veras FP, Pontelli MC, Silva CM, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med 2020:217.
29. Cesta MC, Zippoli M, Marsiglia C, et al. Neutrophil activation and neutrophil extracellular traps (NETs) in COVID-19 ARDS and immunothrombosis. Eur J Immunol 2023;53:e2250010.
30. Zhang R, Sun C, Han Y, et al. Neutrophil autophagy and NETosis in COVID-19: perspectives. Autophagy 2023;19:758-67.
31. Polverino E, Rosales-Mayor E, Dale GE, Dembowsky K, Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest 2017;152:249-62.
32. Castillo MJ, Nakajima K, Zimmerman M, Powers JC. Sensitive substrates for human leukocyte and porcine pancreatic elastase: a study of the merits of various chromophoric and fluorogenic leaving groups in assays for serine proteases. Anal Biochem 1979;99:53-64.
33. Wysocka M, Lesner A, Gruba N, et al. Three wavelength substrate system of neutrophil serine proteinases. Anal Chem 2012;84:7241-8.
34. Sun Q, Li J, Liu WN, Dong QJ, Yang WC, Yang GF. Non-peptide-based fluorogenic small-molecule probe for elastase. Anal Chem 2013;85:11304-11.
35. Aikawa N, Kawasaki Y. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther Clin Risk Manag 2014;10:621-9.
36. Powers JC, Gupton BF, Harley AD, Nishino N, Whitley RJ. Specificity of porcine pancreatic elastase, human leukocyte elastase and cathepsin G. Inhibition with peptide chloromethyl ketones. Biochim Biophys Acta 1977;485:156-66.
37. Jugniot N, Voisin P, Bentaher A, Mellet P. Neutrophil elastase activity imaging: recent approaches in the design and applications of activity-based probes and substrate-based probes. Contrast Media Mol Imaging 2019;2019:7417192.
38. Winiarski Ł, Oleksyszyn J, Sieńczyk M. Human neutrophil elastase phosphonic inhibitors with improved potency of action. J Med Chem 2012;55:6541-53.
39. António JPM, Gonçalves LM, Guedes RC, Moreira R, Gois PMP. Diazaborines as new inhibitors of human neutrophil elastase. ACS Omega 2018;3:7418-23.
40. Zheng Q, Woehl JL, Kitamura S, et al. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc Natl Acad Sci USA 2019;116:18808-14.
41. Xu Y, Qi N, Wen H, Zhang G, Wang Y, Cui H. Synthesis and evaluation of benzenesulfonic acid derivatives as human neutrophil elastase (hNE) inhibitors. Med Chem Res 2021;30:387-98.
42. Al-Horani RA, Aliter KF, Kar S, Mottamal M. Sulfonated nonsaccharide heparin mimetics are potent and noncompetitive inhibitors of human neutrophil elastase. ACS Omega 2021;6:12699-710.
43. Ruivo EF, Gonçalves LM, Carvalho LA, et al. Clickable 4-Oxo-β-lactam-based selective probing for human neutrophil elastase related proteomes. ChemMedChem 2016;11:2037-42.
44. Schulz-Fincke AC, Tikhomirov AS, Braune A, et al. Design of an activity-based probe for human neutrophil elastase: implementation of the lossen rearrangement to induce förster resonance energy transfers. Biochemistry 2018;57:742-52.
45. Anderson BM, Poole DP, Aurelio L, et al. Application of a chemical probe to detect neutrophil elastase activation during inflammatory bowel disease. Sci Rep 2019;9:13295.
46. Liu SY, Xiong H, Li RR, Yang WC, Yang GF. Activity-based near-infrared fluorogenic probe for enabling
47. Ahmad S, Saleem M, Riaz N, et al. The natural polypeptides as significant elastase inhibitors. Front Pharmacol 2020;11:688.
48. Marinaccio L, Stefanucci A, Scioli G, et al. Peptide human neutrophil elastase inhibitors from natural sources: an overview. Int J Mol Sci 2022;23:2924.
49. Wan H, Lee KS, Kim BY, et al. A spider-derived Kunitz-type serine protease inhibitor that acts as a plasmin inhibitor and an elastase inhibitor. PLoS One 2013;8:e53343.
50. Luan N, Zhao Q, Duan Z, et al. Identification and characterization of ShSPI, a Kazal-type elastase inhibitor from the venom of scolopendra hainanum. Toxins (Basel) 2019;11:708.
51. Al-Awadhi FH, Paul VJ, Luesch H. Structural diversity and anticancer activity of marine-derived elastase inhibitors: key features and mechanisms mediating the antimetastatic effects in invasive breast cancer. Chembiochem 2018;19:815-25.
52. Luo D, Luesch H. Ahp-cyclodepsipeptide inhibitors of elastase: lyngbyastatin 7 stability, scalable synthesis, and focused library analysis. ACS Med Chem Lett 2020;11:419-25.
53. Keller L, Canuto KM, Liu C, et al. Tutuilamides A-C: vinyl-chloride-containing cyclodepsipeptides from marine cyanobacteria with potent elastase inhibitory properties. ACS Chem Biol 2020;15:751-7.
54. Crisford H, Sapey E, Stockley RA. Proteinase 3; a potential target in chronic obstructive pulmonary disease and other chronic inflammatory diseases. Respir Res 2018;19:180.
55. Korkmaz B, Lesner A, Guarino C, et al. Inhibitors and antibody fragments as potential anti-inflammatory therapeutics targeting neutrophil proteinase 3 in human disease. Pharmacol Rev 2016;68:603-30.
56. Grzywa R, Lesner A, Korkmaz B, Sieńczyk M. Proteinase 3 phosphonic inhibitors. Biochimie 2019;166:142-9.
57. Witko-Sarsat V, Thieblemont N. Granulomatosis with polyangiitis (Wegener granulomatosis): a proteinase-3 driven disease? Joint Bone Spine 2018;85:185-9.
58. Epinette C, Croix C, Jaquillard L, et al. A selective reversible azapeptide inhibitor of human neutrophil proteinase 3 derived from a high affinity FRET substrate. Biochem Pharmacol 2012;83:788-96.
59. Popow-Stellmaszyk J, Wysocka M, Lesner A, Korkmaz B, Rolka K. A new proteinase 3 substrate with improved selectivity over human neutrophil elastase. Anal Biochem 2013;442:75-82.
60. Guarino C, Legowska M, Epinette C, et al. New selective peptidyl di(chlorophenyl) phosphonate esters for visualizing and blocking neutrophil proteinase 3 in human diseases. J Biol Chem 2014;289:31777-91.
61. Guarino C, Gruba N, Grzywa R, et al. Exploiting the S4-S5 specificity of human neutrophil proteinase 3 to improve the potency of peptidyl di(chlorophenyl)-phosphonate ester inhibitors: a kinetic and molecular modeling analysis. J Med Chem 2018;61:1858-70.
62. Budnjo A, Narawane S, Grauffel C, et al. Reversible ketomethylene-based inhibitors of human neutrophil proteinase 3. J Med Chem 2014;57:9396-408.
63. Hwang TL, Wang WH, Wang TY, Yu HP, Hsieh PW. Synthesis and pharmacological characterization of 2-aminobenzaldehyde oxime analogs as dual inhibitors of neutrophil elastase and proteinase 3. Bioorg Med Chem 2015;23:1123-34.
64. Grzywa R, Burchacka E, Łęcka M, et al. Synthesis of novel phosphonic-type activity-based probes for neutrophil serine proteases and their application in spleen lysates of different organisms. Chembiochem 2014;15:2605-12.
65. Kasperkiewicz P, Altman Y, D'Angelo M, Salvesen GS, Drag M. Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J Am Chem Soc 2017;139:10115-25.
66. Tian S, Swedberg JE, Li CY, Craik DJ, de Veer SJ. Iterative optimization of the cyclic peptide SFTI-1 yields potent inhibitors of neutrophil proteinase 3. ACS Med Chem Lett 2019;10:1234-9.
67. Saidi A, Wartenberg M, Madinier JB, et al. Monitoring human neutrophil activation by a proteinase 3 near-infrared fluorescence substrate-based probe. Bioconjug Chem 2021;32:1782-90.
68. Pagano MB, Bartoli MA, Ennis TL, et al. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci USA 2007;104:2855-60.
69. Hermant B, Bibert S, Concord E, et al. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem 2003;278:14002-12.
70. Ramaha A, Patston PA. Release and degradation of angiotensin I and angiotensin II from angiotensinogen by neutrophil serine proteinases. Arch Biochem Biophys 2002;397:77-83.
71. Okada Y, Nakanishi I. Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett 1989;249:353-6.
72. Chatham WW, Blackburn WD Jr, Heck LW. Additive enhancement of neutrophil collagenase activity by HOCl and cathepsin G. Biochem Biophys Res Commun 1992;184:560-7.
73. Scott FL, Hirst CE, Sun J, Bird CH, Bottomley SP, Bird PI. The Intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood 1999;93:2089-97.
74. Polanowska J, Krokoszynska I, Czapinska H, Watorek W, Dadlez M, Otlewski J. Specificity of human cathepsin G. Biochim Biophys Acta 1998;1386:189-98.
75. Wysocka M, Legowska A, Bulak E, et al. New chromogenic substrates of human neutrophil cathepsin G containing non-natural aromatic amino acid residues in position P(1) selected by combinatorial chemistry methods. Mol Divers 2007;11:93-9.
76. Lesner A, Wysocka M, Guzow K, Wiczk W, Legowska A, Rolka K. Development of sensitive cathepsin G fluorogenic substrate using combinatorial chemistry methods. Anal Biochem 2008;375:306-12.
77. Groborz K, Kołt S, Kasperkiewicz P, Drag M. Internally quenched fluorogenic substrates with unnatural amino acids for cathepsin G investigation. Biochimie 2019;166:103-11.
78. Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 1991;30:485-93.
79. Sieńczyk M, Lesner A, Wysocka M, et al. New potent cathepsin G phosphonate inhibitors. Bioorg Med Chem 2008;16:8863-7.
80. Serim S, Baer P, Verhelst SH. Mixed alkyl aryl phosphonate esters as quenched fluorescent activity-based probes for serine proteases. Org Biomol Chem 2015;13:2293-9.
81. Kam CM, Abuelyaman AS, Li Z, Hudig D, Powers JC. Biotinylated isocoumarins, new inhibitors and reagents for detection, localization, and isolation of serine proteases. Bioconjug Chem 1993;4:560-7.
82. Haedke U, Götz M, Baer P, Verhelst SH. Alkyne derivatives of isocoumarins as clickable activity-based probes for serine proteases. Bioorg Med Chem 2012;20:633-40.
83. Kahler JP, Lenders S, van de Plassche MAT, Verhelst SHL. Facile synthesis of aminomethyl phosphinate esters as serine protease inhibitors with primed site interaction. ACS Med Chem Lett 2020;11:1739-44.
84. Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci USA 2012;109:6229-34.
85. Perera NC, Wiesmüller KH, Larsen MT, et al. NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J Immunol 2013;191:2700-7.
86. Akula S, Thorpe M, Boinapally V, Hellman L. Granule associated serine proteases of hematopoietic cells - an analysis of their appearance and diversification during vertebrate evolution. PLoS One 2015;10:e0143091.
87. Lin SJ, Dong KC, Eigenbrot C, van Lookeren Campagne M, Kirchhofer D. Structures of neutrophil serine protease 4 reveal an unusual mechanism of substrate recognition by a trypsin-fold protease. Structure 2014;22:1333-40.
88. AhYoung AP, Eckard SC, Gogineni A, et al. Neutrophil serine protease 4 is required for mast cell-dependent vascular leakage. Commun Biol 2020;3:687.
89. Kasperkiewicz P, Poreba M, Snipas SJ, et al. Design of a selective substrate and activity based probe for human neutrophil serine protease 4. PLoS One 2015;10:e0132818.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Skoreński M, Torzyk K, Sieńczyk M. Neutrophil serine proteases. Rare Dis Orphan Drugs J 2023;2:6. http://dx.doi.org/10.20517/rdodj.2022.21
AMA Style
Skoreński M, Torzyk K, Sieńczyk M. Neutrophil serine proteases. Rare Disease and Orphan Drugs Journal. 2023; 2(2): 6. http://dx.doi.org/10.20517/rdodj.2022.21
Chicago/Turabian Style
Skoreński, Marcin, Karolina Torzyk, Marcin Sieńczyk. 2023. "Neutrophil serine proteases" Rare Disease and Orphan Drugs Journal. 2, no.2: 6. http://dx.doi.org/10.20517/rdodj.2022.21
ACS Style
Skoreński, M.; Torzyk K.; Sieńczyk M. Neutrophil serine proteases. Rare. Dis. Orphan. Drugs. J. 2023, 2, 6. http://dx.doi.org/10.20517/rdodj.2022.21
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 67 clicks
Cite This Article 67 clicks

 Like This Article 0
likes
Like This Article 0
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.