
Pathogenesis of autoimmune and hereditary pancreatitis with a focus on neutrophil granulocytes and neutrophil serine proteases
 0
0Abstract
Hereditary and autoimmune pancreatitis are two rare forms of inflammatory pancreatic disorders. Both share similarities with acute, acute recurrent, and chronic pancreatitis. Regarding their pathogenesis, the premature activation of the digestive protease trypsinogen and the infiltration of inflammatory cells such as polymorphonuclear leukocytes and macrophages into the pancreas are highly relevant and can reciprocally amplify inflammation. Neutrophil serine proteases are the main components of neutrophil granulocytes and have different pro-inflammatory effects in many diseases. However, their role in pancreatitis is still limited. This section focuses on known findings regarding the role of this group of enzymes in hereditary and autoimmune pancreatitis.
Keywords
INTRODUCTION
Acute pancreatitis is defined as an acute inflammatory condition of the pancreas and is characterized by self-digestion of the pancreatic tissue by its own digestive enzymes. It is one of the most frequent gastrointestinal diseases leading to hospitalization in Europe and the United States, with an estimated annual incidence of 13-45/100,000 inhabitants[1]. Although acute pancreatitis is mostly characterized by a mild course with edematous changes of the organ and complete recovery, a severe disease course occurs in 10%-15% of patients, leading to organ failure and high mortality. Necrotizing pancreatitis is observed in up to 20% of patients and can lead to further complications such as secondary infections requiring further interventions[2]. Alcohol abuse and migrating gallstones are the most common causes of acute pancreatitis and account for approximately 80% of all cases. Less frequent causes include genetic factors, drug-induced effects, anatomic variants, autoimmune diseases, and metabolic disorders such as hypertriglyceridemia or hypercalcemia[3]. This review aims to provide insights into current concepts in the pathophysiology of autoimmune and hereditary pancreatitis by emphasizing the pathogenic function of neutrophil granulocytes and neutrophil serine proteases (NSPs).
HEREDITARY PANCREATITIS
The term hereditary pancreatitis classically refers to mutations in the cationic trypsinogen gene (PRSS1) that are inherited in an autosomal-dominant pattern with high penetrance[4-6]. Patients present with recurrent attacks of acute or chronic pancreatitis without an obvious underlying cause. An autosomal dominant pattern of inheritance was first reported in 1952 by Comfort and Steinberg in a family with chronic pancreatitis[7], and the genetic basis was studied intensively in the following years. A breakthrough was reached in 1996 when mutations in the PRSS1 gene (PRSS1) leading to a gain-of-function were discovered. These led to hereditary pancreatitis[8]. Since 1996, many other genetic variants of PRSS1 have been reported, underlining the pathogenic role of trypsinogen in pancreatitis. More rarely, homozygous mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) and the serine protease inhibitor Kazal-type 1 (SPINK1) genes are linked to hereditary pancreatitis. Other genetic factors increase the risk of recurrent acute or chronic pancreatitis and include variants of carboxypeptidase A1 (CPA1), elastase 3B (CELA3B), and chymotrypsin C (CTRC) genes, as well as heterozygous SPINK1 or CFTR mutations. These variants are considered to have disease-modifying functions together with environmental factors. If acute recurrent or chronic pancreatitis occurs in families with a frequency higher than expected by chance, familial pancreatitis can be suspected. These genes act in a direct or indirect way in the intracellular protease activation cascade and encode proteases and their inhibitors[9,10] [Table 1]. Patients who have hereditary pancreatitis or carry genetic susceptibility factors without Mendelian inheritance patterns mostly suffer from their first acute phase at a significantly younger age compared to patients with other etiologies, and they often have a family history of pancreatitis[11,12]. In addition, a number of genetic mutations are related to faster disease progression and a higher probability of pancreatitis-related complications such as pancreatogenic diabetes (Type 3c), exocrine pancreatic insufficiency, and pancreatic adenocarcinoma[10,11].
Overview of pancreatitis-associated genetic variants
| Gene | Variant | Pathophysiologic change | Publication |
| PRSS1 | R122H | Impaired autolysis of trypsin by mesotrypsin, enzyme Y, and trypsin | Whitcomb et al. 1996 |
| PRSS1 | N29I | Enhanced autoactivation of trypsinogen | Gorry et al. 1997, Sahin-Toth 2000 |
| PRSS1 | E79K | Reduced autoactivation, but increased activation of anionic trypsinogen (PRSS2) | Teich et al. 2004 |
| PRSS1 | R116C | Misfolded trypsin, reduced secretion | Teich et al. 2006, Kereszturi et al. 2009 |
| SPINK1 | L14R | Enhanced intracellular autodigestion, reduced secretion | Kiraly et al. 2007 |
| SPINK1 | L14P | Enhanced intracellular autodigestion, reduced secretion | Kiraly et al. 2007 |
| SPINK1 | N34S | Unknown, probably part of a polygenic model | Witt et al. 2000 |
| CTRC | R254W | Reduced secretion, reduced expression | Rosendahl et al. 2008 |
| CTRC | K247_R254del | No secretion, reduced catalytic activity | Rosendahl et al. 2008 |
| CFTR | Several, mostly in combination with other genetic factors | Reduction of chloride and bicarbonate secretion, detainment of trypsinogen inside of the pancreas | Chang et al. 2007 |
| CPA1 | Several variants | Increased protein misfolding and ER stress | Witt et al. 2014 |
| CEL | CEL-HYB | Increased protein misfolding and ER stress | Fjeld et al. 2015 Fjeld et al. 2022 |
| CELA3B | R90C | Translational upregulation resulting in uncontrolled proteolysis | Moore et al. 2019 |
The best-characterized variants are localized in the cationic trypsinogen gene (PRSS1) and implicate different biochemical reactions: while some mutations are linked to an increase in autocatalytic activation and enhanced stability of active trypsin[4,8,13], others are connected to increased transactivation of anionic trypsinogen[14], the second major trypsinogen isoform, or even protein misfolding, which results in chronic pancreatitis mediated by increased endoplasmic reticulum stress[15].
SPINK1 is an essential part of the protective mechanism against premature and intracellular trypsin activation. The most frequently observed pancreatitis-associated genetic variant is the N34S mutation[16], but no functional change has been elucidated to date. Because this mutation is very common in the general population, it is considered to be more likely a disease-modifying mutation or a component of a polygenic model[17]. Other mutations of the SPINK1 gene are related to reduced secretion of trypsin and increased intracellular autodigestion of acinar cells[18]. Chymotrypsin C (CTRC) forms another part of the protective machinery against intracellular trypsin-dependent digestion. This enzyme is minor human chymotrypsin and promotes the degradation of not only cationic trypsin with high specificity but also the other two trypsinogen isoforms[19]. The two best-characterized CTRC variants related to hereditary pancreatitis are R254W and K247_R254del, both cause reduced secretion and activity of this enzyme[20].
Mutations in the CFTR gene can influence the course and severity of pancreatitis as well. The CFTR protein functions as an adenosine monophosphate (AMP)-dependent chloride channel and regulator of transcellular bicarbonate transport[21-24]. Mutations in the CFTR gene lead to a defective CFTR protein and are associated with cystic fibrosis (CF) in humans, usually with impaired exocrine pancreatic function. Among CF patients without pancreas insufficiency, 20% develop acute recurrent or chronic pancreatitis[25]. The underlying reason for this large number of diseased individuals is that a critical mass of intact pancreatic acinar cells seems to be required for the development of pancreatitis, which is not the case for pancreatic-insufficient patients. More than 1,700 CFTR mutations have already been discovered in CF patients. Despite their high carrier frequency in the population, the occurrence of pancreatitis among those carrying one mutation is extremely low and suggests that multiple interacting factors, i.e. in combination with other mutations in the PRSS1 or SPINK1 genes, are likely to be involved in the pathogenesis of pancreatitis[26].
The entirety of the consequences of CFTR dysfunction in association with pancreatitis remains unknown, but some hypotheses have been advanced. Due to the reduced secretion of bicarbonate in the pancreatic duct, the pH increases and can promote the activation of trypsinogen and the loss of the integrity of cell-cell junctions[27,28]. Carboxyl ester lipase (CEL) is a lipolytic enzyme that is localized in pancreatic acinar cells and lactating mammary glands and is capable of hydrolysis of both water-soluble and water-insoluble lipids[29]. The CEL gene is highly polymorphic and contains a variable number of tandem repeat (VNTR) regions. Single base deletions cause maturity-onset diabetes of the young (MODY). The CEL hybrid allele
Finally, mutations in the carboxypeptidase A1 (CPA1) and elastase 3B (CELA3B) genes have also been found to be associated with hereditary pancreatitis[33,34].
AUTOIMMUNE PANCREATITIS
Autoimmune pancreatitis (AIP) is another rare type of pancreatitis that exhibits features of both acute and chronic pancreatitis. First described by Sarles et al. in 1961, it was termed “autoimmune pancreatitis” in 1995 by Yoshida et al.[35,36]. The exact numbers of incidence and prevalence of AIP are largely unclear; however, they seem to differ among countries. In Germany, the incidence has been reported to be approximately 0.29 per 100,000 people, while in Japan, there is a much higher incidence of 1.4 per 100,000 inhabitants[37,38]. AIP is classified into two subtypes: type 1, also known as lymphoplasmacytic sclerosing pancreatitis (LPSP); and type 2, idiopathic duct-centric chronic pancreatitis (IDCP). Both are associated with increased infiltration of immune cells into the pancreas and are responsive to steroid medication[39].
Each type of AIP has its own characteristics. Hallmarks of type 1 AIP are infiltration by IgG4-positive plasma cells, increased IgG4 titer in the serum, and histological abnormalities such as storiform fibrosis and lymphoplasmacytic infiltration of the pancreatic ducts[40]. In addition to the manifestation in the pancreas, other organs can be involved, including the liver, the biliary tract, the kidneys, and the lungs[41], demonstrating that type 1 AIP is one possible manifestation of the large complex of IgG4-related diseases. Often, the onset of type 1 AIP occurs in patients over 60 years of age, with a predominance of males[42], and is also related to pancreatogenic diabetes[43,44].
In comparison, type 2 AIP is characterized by the infiltration of neutrophils into the pancreatic duct and the surrounding tissue, especially the interlobular duct, with local destruction of the ductal epithelium at what are called granulocytic epithelial lesions (GELs)[45,46]. In comparison to type 1 AIP, type 2 AIP is characterized by a reduction or even complete absence of IgG4-positive plasma cells in the pancreas[46]. No reliable biomarker has been established thus far. Although no other organs are directly involved in the disease, in contrast to what is seen in IgG4-related disorders, type 2 AIP is associated with inflammatory bowel diseases[40]. Patients suffering from type 2 AIP are generally younger, and disease distribution is more gender-balanced[47]. Both types of AIP are associated with different antibodies targeting the plasminogen-binding protein of Helicobacter pylori, the ubiquitin-protein ligase E3 component n-recognin, SPINK1, PRSS1, and PRSS2[48,49]. Because these antibodies are also detectable in other disorders, their value in the diagnosis of AIP is limited.
NEUTROPHIL SERINE PROTEASES IN THE PATHOGENESIS OF PANCREATITIS
Under physiological conditions, pancreatic acinar cells secrete digestive proteases, which are stored as inactive precursors in zymogen granules, into the pancreatic duct. When reaching the duodenum, the serine protease trypsinogen is activated by the duodenal brush border enzyme enterokinase. Active trypsin is able to activate additional trypsinogen and can activate other zymogens secreted into the duodenum, including chymotrypsinogen, proelastase, and procarboxypeptidase A1, A2, and B1, in a cascade-like manner[50]. However, activation of trypsinogen within acinar cells is also possible and is performed either by active trypsin itself or by the lysosomal hydrolase cathepsin B (CTSB). Under normal conditions, several barriers prevent intracellular activation of trypsinogen[51,52], including a shift of intravesical pH to generate an acidic milieu and the presence of inhibiting proteins. One of these proteins is SPINK1, which acts as the major intracellular inhibitor of trypsin[53]. Besides CTSB, the lysosomal hydrolase cathepsin L and chymotrypsin C are also capable of cleaving trypsinogen[20,54]. Moreover, the aspartic protease cathepsin D (CTSD) indirectly acts on trypsinogen as CTSD proteolytically activates pro-CTSB[55]. In contrast, under pathophysiological conditions, the activation of trypsinogen and other downstream proteases occurs inside pancreatic acinar cells, resulting in self-digestion and necrosis of the cells and the release of damage-associated molecular patterns (DAMPs). In addition, the transcription nuclear factor kappa B (NF-κB) is upregulated in pancreatic acinar cells, resulting in the overexpression and secretion of pro-inflammatory cytokines and chemokines[56]. The combination of DAMPs, cytokines, and chemokines augments the activation and infiltration of immune cells into the pancreatic tissue. Activation of trypsinogen occurs not only in acini but also within macrophages that invade the pancreas during acute pancreatitis and phagocytose zymogen-containing vesicles. This process also depends on pH as well as the lysosomal protease CTSB[57]. Neutrophils are a second major immune cell type that invades the pancreas during pancreatitis and can cause local tissue damage in different ways. In rodents with acute pancreatitis in which neutrophils were depleted by anti-neutrophil serum or anti-GR-1-antibodies, disease severity was significantly reduced[58,59,59]. Neutrophils are also able to activate trypsinogen, but the exact underlying mechanism is unknown, although it has been elucidated for macrophages[57]. One study showed that inhibition of matrix metalloproteinase-9 (MMP9) led to a reduction of trypsinogen activation by neutrophils[60], Which was NADPH-mediated[58].
Moreover, neutrophils release NSPs cathepsin G (CTSG), proteinase 3 (PR3), and neutrophil elastase (NE). The cysteine protease cathepsin C (CTSC) seems to have a higher-ranking function, as it activates NSPs[61-63]. Neutrophil serine enzymes are capable of cleaving the tight junction protein E-cadherin, but they differ in their efficiencies. The strongest cleavage effects have been observed for NE. The breakdown of E-cadherin results in a loss of cell-cell connections, facilitating the infiltration of immune cells into pancreatic tissue and the formation of edema[43]. Pharmacological inhibition of NE prevented cleavage of E-cadherin and reduced neutrophil infiltration in the pancreas in vivo[64].In contrast, CTSG and PR3 only have minimal E-cadherin cleavage capabilities in acinar cells, and depletion of CTSG neither decreased trypsinogen activation nor led to an amelioration of pancreatitis severity, although a transient decrease of neutrophil infiltration in the pancreas and extra-pancreatic organs was observed[65]. Furthermore, neutrophils are responsible for the extrusion of neutrophil extracellular traps (NETs), which consist of condensed chromatin and NSPs and can enhance pancreatic damage, promote platelet aggregation, and produce further NSP-associated effects[66]. NETs can also promote trypsinogen activation[67]. The main pathogenetic cellular events in the initiation of acute pancreatitis are summarized in Figure 1.
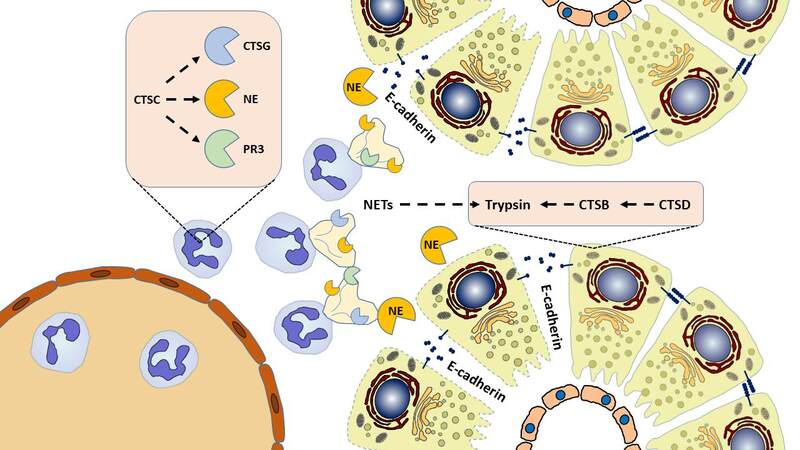
Figure 1. Roles of lysosomal and neutrophil serine proteases in acute pancreatitis. The conversion of trypsinogen into its active form is an important step in the early phase of acute pancreatitis. In addition to autoactivation, trypsinogen is activated by cathepsin B (CTSB) derived from acini and macrophages. Pro-CTSB itself is proteolytically activated by cathepsin D (CTSD). While cathepsin C (CTSC) does not directly act on trypsinogen, it activates the neutrophil serine proteases cathepsin G (CTSG), proteinase 3 (PR3), and neutrophil elastase (NE). NE readily cleaves the cell-adhesion molecule E-cadherin, leading to the dissociation of acini.
The knowledge of the role of neutrophils in general and NSPs in particular in the pathogenesis and course of hereditary and autoimmune pancreatitis remains very limited. The pathogenic role of neutrophils in chronic pancreatitis associated with genetic mutations has been reflected in two murine models. In mice carrying a heterozygous p.D23A mutation in the mouse cationic trypsinogen isoform 7 (T7D23A knock-in), strong auto-activation of trypsinogen was observed, resulting in spontaneous acute and subsequent chronic pancreatitis. Massive infiltration of inflammatory cells including neutrophils and macrophages occurred during the early disease phase. Signs of disease progression were already visible after 4-5 weeks, while immune cells became fewer in number[68]. Transgenic mice expressing the PRSS1 mutant R122H (R122H_mPRSS1) displayed early-onset acinar cell injury and inflammatory cell infiltration, followed by pancreatic fibrosis and an enhanced response to the cholecystokinin analog caerulein[69].
To the best of our knowledge, no currently available literature has specifically focused on the role of NSPs in hereditary pancreatitis, although the organ-injuring effects of neutrophils and elastase in acute pancreatitis are clear in general.
Regarding type 2 AIP, one study compared pancreatic tissue samples of patients with alcoholic chronic pancreatitis, type 1 AIP, and type 2 AIP, and investigated the extent and localization of neutrophilic infiltration. In addition, these authors analyzed the expression of the chemokines interleukin-8 (IL-8) and granulocyte chemotactic protein 2 (GCP-2), both of which can bind to neutrophilic ELR+ CXC receptors and can attract and activate neutrophils. In type 2 AIP, there was higher infiltration of neutrophils into the interlobular ducts and the surrounding tissue, which was absent around the intralobular ducts. In addition, the authors found higher expression of GCP-2 around the interlobular but not around the intralobular ducts, and levels of IL-8 expression remained unaltered. They concluded that the increase of neutrophilic infiltration into the interlobular ducts in type 2 AIP depends on GCP-2 which is secreted by pancreatic epithelial cells[70].
Further knowledge of the role of neutrophils and NSPs in these two rare forms of pancreatitis remains missing. Especially regarding type 2 AIP, knowledge of the interplay between the pancreas, infiltrating neutrophils, and NSPs would be helpful for a more comprehensive understanding of this rare disorder.
MODULATION OF NEUTROPHIL SERINE PROTEASE ACTIVITY
Because NSPs are involved in the pathophysiology of acute pancreatitis, their inhibition is of therapeutic interest. Increased NE activity or an imbalance with endogenous inhibitors can cause a broad range of diseases apart from pancreatitis. Existing NE inhibitors with high inhibitory capacities and low cytotoxicity in vitro have not only been isolated from animal sources and fungi[71] but have also been designed synthetically, thus providing a strong basis for subsequent clinical investigations. The most prominent synthetic compound, AZD9668, has been reported to show beneficial effects in chronic lung diseases[72]. While serine protease inhibitors have attenuated disease severity by reduction of both pancreatic and extra-pancreatic injury in experimental models[73] and prophylactic administration of the serine protease inhibitor gabexate mesylate has reduced the rate of post-ERCP pancreatitis in high-risk patients[74], data regarding usefulness of NSP inhibitors in treating acute pancreatitis are limited and restricted to preclinical models.
Sivelestat, a specific inhibitor of NE, protected against pancreatic and lung injury in rats that had undergone caerulein- or taurocholate-induced acute pancreatitis[75]. Additive effects were achieved when treatment was combined with antioxidant resveratrol[76]. Lex032, an inhibitor of elastase and cathepsin G, ameliorated microcirculatory function in ischemia/reperfusion-induced acute pancreatitis by the preservation of capillary perfusion, absence of interleukin-6 increase, and preservation of mean arterial pressure during reperfusion time, indicating usefulness in preventing pancreatic tissue damage from ischemia/reperfusion[77]. Despite promising results in preclinical investigations, the effectiveness of NSP modulation in clinical use remains to be validated.
CONCLUSION
Despite advances in understanding the pathogenesis of acute pancreatitis, the precise cellular and subcellular mechanisms of the early disease phase are not entirely understood. Neutrophils are recruited to inflamed sites and drive inflammatory reactions by several processes, including the activation of NSPs. Several animal models suggest a favorable effect on acute pancreatitis when neutrophil elastase is inhibited. Recent data support the pro-inflammatory role of NSPs in acute pancreatitis; however, their pathogenic functions in autoimmune and hereditary pancreatitis, two rarer forms of pancreatitis, still need to be clarified.
DECLARATIONS
Authors’ contributionsLiterature research and writing: Zierke L, Gischke M, Tran QT, Aghdassi AA
Final approval of the draft: Zierke L, Gischke M, Tran QT, Aghdassi AA
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was funded by the Deutsche Forschungsgemeinschaft (DFG AG 203/4-1).
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Peery AF, Crockett SD, Murphy CC, et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2018. Gastroenterology 2019;156:254-272.e11.
2. Wiese ML, Urban S, von Rheinbaben S, et al. Identification of early predictors for infected necrosis in acute pancreatitis. BMC Gastroenterol 2022;22:405.
3. Weiss FU, Laemmerhirt F, Lerch MM. Acute pancreatitis: genetic risk and clinical implications. J Clin Med 2021;10:190.
4. Gorry MC, Gabbaizedeh D, Furey W, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology 1997;113:1063-8.
6. Girodon E, Rebours V, Chen JM, et al. Clinical interpretation of
7. Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 1952;21:54-63.
8. Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141-5.
9. Coté GA, Yadav D, Slivka A, et al. North American Pancreatitis Study Group. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 2011;9:266-73; quiz e27.
10. Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut 2009;58:97-103.
11. Howes N, Lerch MM, Greenhalf W, et al. European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004;2:252-61.
12. Sultan M, Werlin S, Venkatasubramani N. Genetic prevalence and characteristics in children with recurrent pancreatitis. J Pediatr Gastroenterol Nutr 2012;54:645-50.
13. Sahin-Tóth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem 2000;275:22750-5.
14. Teich N, Le Maréchal C, Kukor Z, et al. Interaction between trypsinogen isoforms in genetically determined pancreatitis: mutation E79K in cationic trypsin (PRSS1) causes increased transactivation of anionic trypsinogen (PRSS2). Hum Mutat 2004;23:22-31.
15. Kereszturi E, Szmola R, Kukor Z, et al. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat 2009;30:575-82.
16. Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000;25:213-6.
17. Schneider A. Serine protease inhibitor kazal type 1 mutations and pancreatitis. Clin Lab Med 2005;25:61-78.
18. Király O, Boulling A, Witt H, et al. Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum Mutat 2007;28:469-76.
19. Szmola R, Sahin-Tóth M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht’s enzyme Y. Proc Natl Acad Sci USA 2007;104:11227-32.
20. Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (
21. Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Generation of cAMP-activated chloride currents by expression of CFTR. Science 1991;251:679-82.
23. Reddy MM, Quinton PM. Selective activation of cystic fibrosis transmembrane conductance regulator CL- and HCO3- conductances. JOP 2001;2(4 Suppl):212-8.
24. Stutts MJ, Canessa CM, Olsen JC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science 1995;269:847-50.
25. Durno C, Corey M, Zielenski J, Tullis E, Tsui LC, Durie P. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 2002;123:1857-64.
26. Ooi CY, Dorfman R, Cipolli M, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011;140:153-61.
27. Behrendorff N, Floetenmeyer M, Schwiening C, Thorn P. Protons released during pancreatic acinar cell secretion acidify the lumen and contribute to pancreatitis in mice. Gastroenterology 2010;139:1711-20, 1720.e1.
28. Pallagi P, Venglovecz V, Rakonczay Z Jr, et al. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl⁻ channels and luminal anion exchangers. Gastroenterology 2011;141:2228-2239.e6.
29. Hui DY, Howles PN. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J Lipid Res 2002;43:2017-30.
30. Fjeld K, Weiss FU, Lasher D, et al. A recombined allele of the lipase gene
31. Fjeld K, Gravdal A, Brekke RS, et al. The genetic risk factor
32. El Jellas K, Dušátková P, Haldorsen IS, et al. Two new mutations in the
33. Witt H, Beer S, Rosendahl J, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 2013;45:1216-20.
34. Moore PC, Cortez JT, Chamberlain CE, et al. Elastase 3B mutation links to familial pancreatitis with diabetes and pancreatic adenocarcinoma. J Clin Invest 2019;129:4676-81.
35. SARLES H, SARLES JC, MURATORE R, GUIEN C. Chronic inflammatory sclerosis of the pancreas-an autonomous pancreatic disease? Am J Dig Dis 1961;6:688-98.
36. Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995;40:1561-8.
37. Schneider A, Michaely H, Weiss C, et al. Prevalence and incidence of autoimmune pancreatitis in the population living in the southwest of Germany. Digestion 2017;96:187-98.
38. Kanno A, Masamune A, Okazaki K, et al. Research Committee of Intractable Diseases of the Pancreas. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2011. Pancreas 2015;44:535-9.
39. Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003;27:235-8.
40. Kamisawa T, Chari ST, Giday SA, et al. Clinical profile of autoimmune pancreatitis and its histological subtypes: an international multicenter survey. Pancreas 2011;40:809-14.
41. Sandrasegaran K, Menias CO. Imaging in autoimmune pancreatitis and immunoglobulin g4-related disease of the abdomen. Gastroenterol Clin North Am 2018;47:603-19.
42. Hart PA, Kamisawa T, Brugge WR, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013;62:1771-6.
43. John DS, Aschenbach J, Krüger B, et al. Deficiency of cathepsin C ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of E-cadherin. J Biol Chem 2019;294:697-707.
44. Ito T, Nakamura T, Fujimori N, et al. Characteristics of pancreatic diabetes in patients with autoimmune pancreatitis. J Dig Dis 2011;12:210-6.
45. Zamboni G, Lüttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004;445:552-63.
46. Shinagare S, Shinagare AB, Deshpande V. Autoimmune pancreatitis: a guide for the histopathologist. Semin Diagn Pathol 2012;29:197-204.
47. Ikeura T, Manfredi R, Zamboni G, et al. Application of international consensus diagnostic criteria to an Italian series of autoimmune pancreatitis. United European Gastroenterol J 2013;1:276-84.
48. Löhr JM, Faissner R, Koczan D, et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol 2010;105:2060-71.
49. Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009;361:2135-42.
50. Mayerle J, Sendler M, Hegyi E, Beyer G, Lerch MM, Sahin-Tóth M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 2019;156:1951-1968.e1.
51. Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 2000;106:773-81.
52. Saluja A, Hashimoto S, Saluja M, Powers RE, Meldolesi J, Steer ML. Subcellular redistribution of lysosomal enzymes during caerulein-induced pancreatitis. Am J Physiol 1987;253:G508-16.
53. Ohmuraya M, Yamamura K. Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Exp Anim 2011;60:433-44.
54. Wartmann T, Mayerle J, Kähne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 2010;138:726-37.
55. Aghdassi AA, John DS, Sendler M, et al. Cathepsin D regulates cathepsin B activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J Biol Chem 2018;293:1018-29.
56. Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol 1998;275:G1402-14.
57. Sendler M, Weiss FU, Golchert J, et al. Cathepsin B-mediated activation of trypsinogen in endocytosing macrophages increases severity of pancreatitis in mice. Gastroenterology 2018;154:704-718.e10.
58. Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002;122:974-84.
59. Abdulla A, Awla D, Thorlacius H, Regnér S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol 2011;90:975-82.
60. Awla D, Abdulla A, Syk I, Jeppsson B, Regnér S, Thorlacius H. Neutrophil-derived matrix metalloproteinase-9 is a potent activator of trypsinogen in acinar cells in acute pancreatitis. J Leukoc Biol 2012;91:711-9.
61. Mcguire M, Lipsky P, Thiele D. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. Journal of Biological Chemistry 1993;268:2458-67.
62. Turk D, Janjić V, Stern I, et al. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J 2001;20:6570-82.
63. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002;109:363-71.
64. Mayerle J, Schnekenburger J, Krüger B, et al. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology 2005;129:1251-67.
65. Aghdassi AA, John DS, Sendler M, et al. Absence of the neutrophil serine protease cathepsin G decreases neutrophil granulocyte infiltration but does not change the severity of acute pancreatitis. Sci Rep 2019;9:16774.
66. Leppkes M, Maueröder C, Hirth S, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun 2016;7:10973.
67. Merza M, Hartman H, Rahman M, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology 2015;149:1920-1931.e8.
68. Geisz A, Sahin-Tóth M. A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation. Nat Commun 2018;9:5033.
69. Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 2006;131:1844-55.
70. Mitsuyama T, Uchida K, Sumimoto K, et al. Comparison of neutrophil infiltration between type 1 and type 2 autoimmune pancreatitis. Pancreatology 2015;15:271-80.
71. Marinaccio L, Stefanucci A, Scioli G, et al. Peptide human neutrophil elastase inhibitors from natural sources: an overview. Int J Mol Sci 2022;23:2924.
72. Sjö P. Neutrophil elastase inhibitors: recent advances in the development of mechanism-based and nonelectrophilic inhibitors. Future Med Chem 2012;4:651-60.
73. Cao C, Yin C, Shou S, et al. Ulinastatin protects against LPS-induced acute lung injury by attenuating TLR4/NF-κB pathway activation and reducing inflammatory mediators. Shock 2018;50:595-605.
74. Testoni PA, Mariani A, Masci E, Curioni S. Frequency of post-ERCP pancreatitis in a single tertiary referral centre without and with routine prophylaxis with gabexate: a 6-year survey and cost-effectiveness analysis. Dig Liver Dis 2006;38:588-95.
75. Cao J, Liu Q. Protective effects of sivelestat in a caerulein-induced rat acute pancreatitis model. Inflammation 2013;36:1348-56.
76. Wang H, Wang S, Tang A, Gong H, Ma P, Chen L. Combined effects of sivelestat and resveratrol on severe acute pancreatitis-associated lung injury in rats. Exp Lung Res 2014;40:288-97.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zierke L, Gischke M, Tran QT, Aghdassi AA. Pathogenesis of autoimmune and hereditary pancreatitis with a focus on neutrophil granulocytes and neutrophil serine proteases. Rare Dis Orphan Drugs J 2023;2:10. http://dx.doi.org/10.20517/rdodj.2022.17
AMA Style
Zierke L, Gischke M, Tran QT, Aghdassi AA. Pathogenesis of autoimmune and hereditary pancreatitis with a focus on neutrophil granulocytes and neutrophil serine proteases. Rare Disease and Orphan Drugs Journal. 2023; 2(2): 10. http://dx.doi.org/10.20517/rdodj.2022.17
Chicago/Turabian Style
Zierke, Lukas, Marcel Gischke, Quang Trung Tran, Ali A. Aghdassi. 2023. "Pathogenesis of autoimmune and hereditary pancreatitis with a focus on neutrophil granulocytes and neutrophil serine proteases" Rare Disease and Orphan Drugs Journal. 2, no.2: 10. http://dx.doi.org/10.20517/rdodj.2022.17
ACS Style
Zierke, L.; Gischke M.; Tran QT.; Aghdassi AA. Pathogenesis of autoimmune and hereditary pancreatitis with a focus on neutrophil granulocytes and neutrophil serine proteases. Rare. Dis. Orphan. Drugs. J. 2023, 2, 10. http://dx.doi.org/10.20517/rdodj.2022.17
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 1
likes
Like This Article 1
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.