
Biallelic cubilin pathogenic variants as a cause of « benign » proteinuria: implications for clinical management
 0
0Abstract
The recent description of a cohort with both adults and children harboring biallelic pathogenic variants of CUBN changed the paradigm of the management of isolated proteinuria. Indeed, the detection of proteinuria in a patient, regardless of age, often leads to an exhaustive check-up including kidney biopsy but also the prescription of renin-angiotensin system (RAS) blockers to slow the progression of kidney disease. Patients with CUBN variants have nondetrimental proteinuria and are non-responsive to RAS blockers. We herein describe 2 siblings treated for isolated proteinuria for several years, eventually diagnosed with CUBN biallelic pathogenic variants (c.703 C > T and c.10363-3A > G). We review the physio-pathological mechanisms of this newly discovered disease and discuss implications for clinical management.
Keywords
INTRODUCTION
Discovering isolated proteinuria in a patient should lead to an appropriate work-up, including biological analysis, kidney ultrasound, and sometimes kidney biopsy. Proteinuria may be glomerular (mainly albuminuria with or without microscopic hematuria), tubular (low molecular weight proteins such as α1-microglobulin, retinol-binding protein, or β2-microglobulin), or both. Regardless of the cause of proteinuria, blockers of the renin-angiotensin (RAS) system, such as angiotensin receptor blockers or angiotensin-converting enzyme (ACE) inhibitors, are usually prescribed to decrease proteinuria and therefore slow down kidney damage. Indeed, proteinuria is a well-established risk factor for progressive kidney disease[1,2]. Increased glomerular capillary pressure may induce excessive filtration of plasma proteins and podocyte dysfunction. The increased reabsorption of plasma proteins by proximal tubular cells may be toxic and lead to their apoptosis and to interstitial fibrosis[1,3]. This paradigm recently changed with the discovery of pathogenic variants in the cubilin (CUBN) gene[4] that may cause nondetrimental proteinuria. We report the case of 2 siblings with such variants and discuss their clinical management.
CLINICAL CASE
An 8-year-old girl of Turkish origin with no relevant medical history presented to the Pediatric Nephrology clinic for nocturnal enuresis. An initial check-up revealed isolated mild proteinuria (protein/creatinine ratio 1.2 g/g) made up of albuminuria. The estimated glomerular filtration rate (eGFR) was normal as was the ultrasound of the kidneys. A kidney biopsy was performed but only contained a single glomerulus with a normal aspect by light microscopy. Immunofluorescence microscopy was negative. Enalapril was started at 2.5 mg/day. She remained on this medication for 15 years and proteinuria remained stable at 0.7 g/g to 1.2 g/g despite an increase in enalapril dosage up to 15 mg/day. Enalapril was poorly tolerated, with frequent episodes of orthostatic hypotension. eGFR remained completely normal.
Her younger brother was also diagnosed with moderate isolated proteinuria (0.9 g/g) during a check-up for hyperactive bladder. His kidney biopsy showed eight normal glomeruli by light microscopy. Immunofluorescence microscopy was negative. Enalapril was started and proteinuria remained stable at 0.6-0.9 g/g during 8 years with preserved eGFR. Both parents have normal urinalysis without proteinuria. The first genetic testing was performed in 2018 in both siblings with a panel including 20 genes related to proteinuria. Results were not contributive as a single variant of unknown origin (VUS) of the ACTN4 gene was found in the sister but not in her brother. Updated genetic testing performed in 2022 (NGS panel enriched in 40genes important for proteinuric renal diseases)[5] revealed in both siblings the presence of a pathogenic nonsense variant (ACMG/AMP class 5) together with a potential splice disrupting variant (ACMG/AMP class 3) in the CUBN (NM_001081.4):c.703 C > T (p.Arg235ter) and c.10363-3A > G (p.?). Cubulin (CUBN) is dominated by 27 contiguous CUB domains, and variants occurring within the C-terminal (after the CUB8 domain) are usually associated with isolated proteinuria. Here, p.R235* led to truncation before CUB8, but none of the patients presented with low levels of vitamin B12 and megaloblastic anemia Enalapril was therefore discontinued in both patients.
DISCUSSION
The proximal tubule reabsorbs large amounts of low-molecular-weight proteins but also albumin and electrolytes from the glomerular filtrate. Megalin (LRP2), cubilin (CUBN), and amnionless protein (AMN) are located in the apical part of proximal tubular cells and are responsible for receptor-mediated endocytosis of proteins filtered through the glomerular barrier[3]. Cubilin has been shown to have an essential role in albumin reabsorption and is encoded by the CUBN gene [Figure 1][6]. Biallelic pathogenic variants in the CUBN gene cause Imerslund-Gräsbeck syndrome (OMIM 261100), also called selective vitamin B12 (cobalamin) malabsorption with proteinuria[6]. In this syndrome resulting in megaloblastic anemia responsive to parenteral vitamin B12 therapy, half of the patients present with mild proteinuria and normal eGFR. The mechanism of megaloblastic anemia is a defect in the receptor of the vitamin B12-intrinsic factor complex in the ileal enterocyte. CUBN and AMN proteins represent the two subunits of this receptor. In patients with Imerslund-Gräsbeck syndrome, proteinuria persists over decades[7,8]. Most CUBN pathogenic variants are located in the N-terminal half of the cubilin gene [Figure 2].
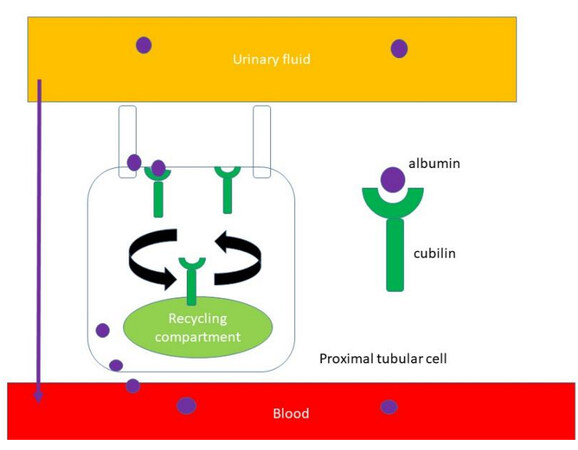
Figure 1. Albumin reabsorption in proximal tubular cells. Cubilin is located in the apical part of proximal tubular cells and is responsible for receptor-mediated endocytosis of albumin filtered through the glomerular barrier. After the decoupling of cubulin-albumin ligation, albumin is then released from the basolateral cell surface into the circulation.
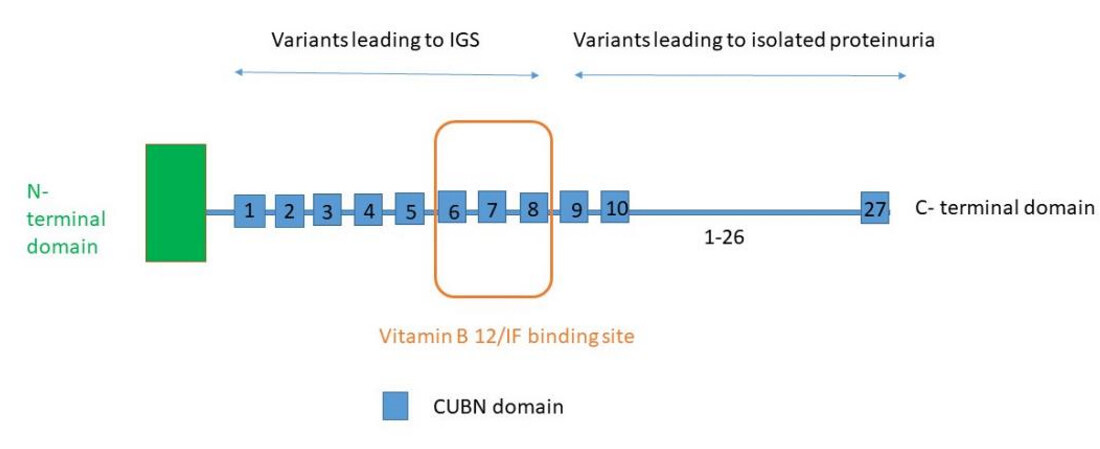
Figure 2. Cubilin protein structure. Cubilin (CUB) is a 460 kDa glycoprotein without transmembrane domain. It acts as a receptor for intrinsic factor-vitamin B12 complexes. There are 27 CUB domains. Intrinsic factor and vitamin B12 binding region is located in domains 5 to 8. The precise location of the binding sites for albumin remains unclear, but it is supposed to be located in the CUB domains near the C-terminal area. IGS : Imerslund-Grasbëck syndrome; IF : intrinsic factor.
Recently, bilallelic pathogenic variants in the C-terminal domain of CUBN were described as leading to isolated chronic proteinuria[4]. Indeed, Bedin et al. identified 39 patients with biallelic CUBN variants among 2216 individuals with suspected genetic kidney disease including proteinuric patients. Proteinuria ranged from 0,5-3 g/day with an average age at discovery of 10.9 years. When measured, albuminuria represented more than half of proteinuria and β2-microglobulin urine level was low or absent. Kidney biopsies were available in 19 patients and did not show any specific lesion in 11 patients. Four kidney biopsies had electronic microscopy (EM) evaluation, two were normal and two revealed glomerular synechiae. The use of ACE inhibitors did not lower proteinuria which remained stable over years. eGFR was normal in all patients, even those older than 50 years. Bedin et al. also identified a phenotype-genotype correlation. Indeed, variants located after the CUB8 domain (included in the vitamin B12/intrinsic factor binding region) lead to isolated proteinuria, whereas variants located before the CUB8 domain lead to Imerslund-Gräsbeck syndrome, a finding suggesting that there are separate binding sites in cubilin for vitamin B12–intrinsic factor (VitB12-IF) and albumin but the precise location of the binding sites for albumin remains unclear [Figure 2]. The latter should thus bind to more carboxy-terminal CUB domains. However, the isolated proteinuria caused by the p.R235* variant located before CUB8 and leading to premature truncation of cubilin illustrates how it remains complex to determine with certainty the phenotype. In addition, four specific C-terminal variants previously showed strong associations with albuminuria in GWAS[9-13]. These CUBN variants were associated with higher eGFR in Bedin et al. study[4].
In another recent cohort, Domingo-Gallego et al.[14] identified 15 patients with mild proteinuria (0.5-1.8 g/day) having homozygous or compound heterozygous pathogenic variants in the C-terminal CUBN protein. In most cases, proteinuria was detected incidentally, as in our patients. They confirmed the glomerular nature of proteinuria, normal kidney histology, lack of response to RAS blockade, and preserved eGFR in adulthood. Six children from Turkey were also identified with biallelic CUBN pathogenic variants located at the C-terminal domain of the protein[15]. One child had a second kidney biopsy 3 years after the first normal kidney biopsy. This second biopsy revealed one periglomerular fibrosis among 27 glomeruli. Yang et al.[16] also reported glomerulosclerosis and effacement of foot processes in podocytes on electronic microscopy (EM) in three children with CUBN pathogenic variants. The authors suggested a role of podocyte dysfunction together with a defect in the re-uptake of albumin in the proximal tubule of patients with CUBN mutations. These data need to be confirmed. Indeed, these structural changes in podocytes were absent in two children on EM[17]. These authors showed that CUBN variants induce changes in the scaffolding capabilities of cubilin protein in vitro. These changes reduce the interactions between CUB and AMN, leading to an aberrant localization of AMN in the cytoplasm of proximal tubular cells instead of the cell membrane. This may interrupt the receptor-mediated endocytosis that re-uptakes filtered albumin.
In conclusion, in the absence of functional cubilin in proximal tubular cells, albumin reabsorption is incomplete and this leads to mild albuminuria. This mechanism acts downstream of the glomerular barrier and does not affect the intraglomerular pressure and is thus not expected to damage podocytes. This explains why anti-proteinuric agents such as ACE inhibitors do not succeed in lowering this particular proteinuria. Detection of CUBN pathogenic variants is crucial in clinical nephrology because it prevents unnecessary kidney biopsies but also the use of RAS blockers and their potential side effects such as symptomatic hypotension or rarely angioneurotic edema. The benign course of this disease needs to be confirmed by a longer follow-up.
DECLARATIONS
Authors’ contributionsContributed to the concept, design, draft, and revision of this manuscript: Gillion V, Dahan K, Godefroid N
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Roscioni SS, Lambers Heerspink HJ, de Zeeuw D. Microalbuminuria: target for renoprotective therapy PRO. Kidney Int 2014;86:40-9.
2. Hemmelgarn BR, Manns BJ, Lloyd A, et al. Alberta Kidney Disease Network. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 2010;303:423-9.
3. Quinlan C. CUBN variants uncouple proteinuria from kidney function. Nat Rev Nephrol 2020;16:135-6.
4. Bedin M, Boyer O, Servais A, et al. Human C-terminal CUBN variants associate with chronic proteinuria and normal renal function. J Clin Invest 2020;130:335-44.
5. Knoers N, Antignac C, Bergmann C, et al. Genetic testing in the diagnosis of chronic kidney disease: recommendations for clinical practice. Nephrol Dial Transplant 2022;37:239-54.
6. Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol; 2010. 1859-67
7. Gräsbeck R. Imerslund-Gräsbeck syndrome (selective vitamin B(12) malabsorption with proteinuria). Orphanet J Rare Dis 2006;1:17.
8. Aminoff M, Carter JE, Chadwick RB, et al. Mutations in CUBN, encoding the intrinsic factor-vitamin B12 receptor, cubilin, cause hereditary megaloblastic anaemia 1. Nat Genet 1999;21:309-13.
9. Ahluwalia TS, Schulz CA, Waage J, et al. A novel rare CUBN variant and three additional genes identified in Europeans with and without diabetes: results from an exome-wide association study of albuminuria. Diabetologia 2019;62:292-305.
10. Böger CA, Chen MH, Tin A, et al. CKDGen Consortium. CUBN is a gene locus for albuminuria. J Am Soc Nephrol 2011;22:555-70.
11. Haas ME, Aragam KG, Emdin CA, et al. International Consortium for Blood Pressure. Genetic association of albuminuria with cardiometabolic disease and blood pressure. Am J Hum Genet 2018;103:461-73.
12. Zanetti D, Rao A, Gustafsson S, Assimes TL, Montgomery SB, Ingelsson E. Identification of 22 novel loci associated with urinary biomarkers of albumin, sodium, and potassium excretion. Kidney Int 2019;95:1197-208.
13. Teumer A, Tin A, Sorice R, et al. DCCT/EDIC. Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes 2016;65:803-17.
14. Domingo-Gallego A, Pybus M, Madariaga L, et al. Clinical and genetic characterization of a cohort of proteinuric patients with biallelic CUBN variants. Nephrol Dial Transplant 2022;37:1906-15.
15. Cicek N, Alpay H, Guven S, et al. Clinical and genetic characterization of children with cubilin variants. Pediatr Nephrol 2023;38:1381-5.
16. Yang J, Xu Y, Deng L, et al. CUBN gene mutations may cause focal segmental glomerulosclerosis (FSGS) in children. BMC Nephrol 2022;23:15.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Gillion V, Dahan K, Godefroid N. Biallelic cubilin pathogenic variants as a cause of « benign » proteinuria: implications for clinical management. Rare Dis Orphan Drugs J 2023;2:11. http://dx.doi.org/10.20517/rdodj.2022.23
AMA Style
Gillion V, Dahan K, Godefroid N. Biallelic cubilin pathogenic variants as a cause of « benign » proteinuria: implications for clinical management. Rare Disease and Orphan Drugs Journal. 2023; 2(3): 11. http://dx.doi.org/10.20517/rdodj.2022.23
Chicago/Turabian Style
Gillion, Valentine, Karin Dahan, Nathalie Godefroid. 2023. "Biallelic cubilin pathogenic variants as a cause of « benign » proteinuria: implications for clinical management" Rare Disease and Orphan Drugs Journal. 2, no.3: 11. http://dx.doi.org/10.20517/rdodj.2022.23
ACS Style
Gillion, V.; Dahan K.; Godefroid N. Biallelic cubilin pathogenic variants as a cause of « benign » proteinuria: implications for clinical management. Rare. Dis. Orphan. Drugs. J. 2023, 2, 11. http://dx.doi.org/10.20517/rdodj.2022.23
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 1
likes
Like This Article 1
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.