
Cathepsin C: structure, function, and pharmacological targeting
 0
0
Abstract
Cathepsin C is a papain-like cysteine peptidase known primarily for its involvement in the activation of serine peptidases in neutrophils and other immune cells. Its critical role in this process qualifies cathepsin C as a target for the treatment of inflammatory diseases, and its most advanced inhibitor, brensocatib (Insmed), is currently in phase 3 clinical trials for the treatment of non-cystic fibrosis bronchiectasis. Beyond neutrophils, its importance is highlighted by loss-of-function mutations that cause the recessively inherited Papillon-Lefèvre syndrome. At the molecular level, cathepsin C has several structural and functional features that set it apart from other members of the family and enable its selective targeting. It possesses dipeptidyl-peptidase activity (its other common name is dipeptidyl-peptidase I) due to the presence of an additional exclusion domain that also controls its stepwise tetramerization during maturation. In this review article, we summarize the current state of the art regarding the biochemical properties of cathepsin C, its physiological and pathological roles in neutrophils and beyond, and recent advances in the development and evaluation of cathepsin C inhibitors.
Keywords
INTRODUCTION
Cathepsin C (also known as dipeptidyl-peptidase I, EC 3.4.14.1) was among the first peptidases identified in animal tissues[1]. It belongs to the papain-like family of cysteine peptidases (PLPs), which is classified as family C1, clan CA in the MEROPS database of peptidases, inhibitors, and substrates[2]. It is found in animals and several other eukaryote lineages[3,4]. In humans, it is encoded by the CATC gene located at chromosomal location 11q14.2. It is ubiquitously expressed in humans and other mammals[5] and is a unique peptidase both in terms of its biochemical properties and its specific physiological and pathological functions[3]. It plays a pivotal role in the activation of effector serine peptidases in immune cells such as neutrophils, cytotoxic T lymphocytes, and others. This led to the establishment of cathepsin C as a promising target for the treatment of inflammatory diseases that are characterized by excessive activation of cells of the immune system[6]. Numerous more or less successful inhibitors of cathepsin C have been synthesized and evaluated to date[6]. The most advanced inhibitor, brensocatib (Insmed, Inc, USA), is currently undergoing a phase 3 clinical trial for the treatment of non-cystic fibrosis bronchiectasis[7]. Furthermore, cathepsin C is well-known for the fact that mutations in the CATC gene cause the recessive hereditary diseases called Papillon-Lefèvre syndrome and Haim-Munk syndrome[8-10], highlighting the systemic effects of cathepsin C deficiency. Symptoms include early periodontitis, pathological thickening of the skin on the palms and feet, and increased susceptibility to infection[11]. Apart from these, many other (patho)physiological functions have been attributed to cathepsin C in recent decades.
In this review, we summarize the current state of the art on the structural and functional properties of cathepsin C, its processing and trafficking in the cell, and its roles in human physiology and pathology. As this review is part of the special issue “Neutrophil Serine Proteases in Rare Diseases”, the primary focus is on its roles in the immune system, particularly in neutrophils. In the end, we also provide an overview of current and emerging strategies for the pharmacological targeting of cathepsin C in human disease.
STRUCTURE AND FUNCTION OF CATHEPSIN C
Many expert reviews on the structural and functional properties of PLPs in general are available, e.g., references[3,12], and the reader is advised to refer to these for more in-depth information on this topic. Herein, we are focusing on cathepsin C and its specific structural and functional properties. Like other members of the PLP family, cathepsin C is synthesized as an inactive precursor called procathepsin C. Its active site contains a Cys-His catalytic dyad that forms a thiolate-imidazolium ion pair (residues Cys234 and His381 according to procathepsin C numbering). What distinguishes it from other PLPs is the additional exclusion domain, the fold of which is similar to that of metalloprotease inhibitors[13]. The exclusion domain is located at the N-terminus of procathepsin C and is separated from the catalytic peptidase domain by the propeptide [Figure 1A]. Upon activation, the propeptide (residues Ala111 through His206) is removed, leaving the exclusion domain non-covalently bound to the peptidase domain, which in turn is cleaved into a heavy chain and a light chain by cleavage between Arg370 and Asp371[13]. As its name suggests, the crucial functional role of the exclusion domain is that it sterically hinders access to the active site beyond site S2 [Figure 1B], making cathepsin C a dipeptidyl-peptidase, i.e., an exopeptidase that cleaves dipeptides of the N-termini of its substrates. Moreover, the side chain of residue Asp1 interacts with the N-terminus of the substrate and stabilizes its binding[13]. The selectivity of the S2 binding site, which is the primary specificity determinant of PLPs, is dominated by two negative charges, namely the aforementioned side chain of residue Asp1 at the entrance of the pocket and a chloride ion at the bottom of a deep hydrophobic pocket [Figure 1C]. The latter is required for enzyme activity and is another distinguishing feature of cathepsin C that sets it apart from other PLPs[13,14]. In our recent evolutionary analysis of cathepsin C, we found that residue Asp1 is strictly conserved in all cathepsin C enzymes throughout the phylogenetic tree, suggesting that dipeptidyl-peptidase activity is the conserved enzymatic activity of these enzymes in all species[4]. The substrate binding sites S1, S1', and S2' do not have any special features compared to other members of the family[15]. Cathepsin C has a relatively broad substrate specificity[16-19] and usually acts by the stepwise removal of dipeptides from the N-terminus of the protein substrate. It stops only in three cases: when the N-terminal amino group is blocked, when a proline residue is adjacent to the cleavage site (P1 or P1’ position), or when the N-terminal residues are lysine or arginine. Indeed, derivatives of positively charged and aromatic amino acids have been found to competitively inhibit cathepsin C[20]. Nevertheless, the enzyme shows specific preferences for certain residues at positions P1 and P2[16-18] and at positions P1’ and P2’[19]. Due to its activity, most substrates used to measure cathepsin C activity in vitro are synthetic dipeptide derivatives. The first reported substrates were Gly-Phe-p-nitroanilide (Gly-Phe-pNA) and Gly-Phe-β-naphthylamide (Gly-Phe- βNA)[15]. Their 7-amido-4-methylcoumarin analog (Gly-Phe-AMC) is the most commonly used fluorogenic substrate in the published literature, although better substrates containing natural and unnatural amino acids, such as L-norleucine, L-4-benzoylphenylalanine or L-glutamic acid benzyl ester, have been identified by library screening[16-18]. In addition, highly specific internally quenched substrates have been developed for the detection of cathepsin C activity in biological samples[19]. Like most PLPs, cathepsin C shows optimal activity under slightly acidic pH conditions[21,22]. Unlike others, it also requires chloride (or other halides) ions for optimal activity below pH 7[14,23]. At neutral and basic pH, cathepsin C also exhibits transferase activity[24,25], but similar activity has been observed with other related and unrelated peptidases, e.g., papain[26]. While most cysteine cathepsins are monomers, mature human cathepsin C is a homotetramer organized as a dimer of dimers[13,22] [Figure 1D]. The exclusion domain plays an indispensable role in tetramer formation, as each copy interacts with the peptidase domains of two adjacent subunits. In the tetramer, the active sites of all subunits are fully exposed to the solvent, indicating that all four subunits can function simultaneously. Interestingly, distant cathepsin C homologs have been identified in unicellular eukaryotes, e.g., plasmodium, which were shown to be monomers[16]. We have recently conducted a phylogenetic and computational analysis of the evolution of cathepsin C and found that the tetrameric form is likely restricted to animal cathepsin C homologs[4].

Figure 1. Structural characteristics of cathepsin C. (A) Schematic representation of the (pro)cathepsin C structure; (B) Schematic representation of the active site of papain-like peptidases adapted for cathepsin C. The substrate binding sites on the enzyme are labeled S3 through S2'. The substrate is presented with white circles corresponding to individual residues and residues P2 through P2' are labelled. The arrow denotes the scissile bond; (C) Three-dimensional structure of a cathepsin C subunit. The peptidase domain is shown in molecular surface representation and the exclusion domain in cartoon representation. Active site residues Cys234 and His381 are colored yellow and blue, respectively, Asp1 is shown as red sticks, and the chloride ion cofactor as a green sphere; (D) The cathepsin C tetramer shown in cartoon representation. Exclusion domains are colored blue and peptidase domains are colored tan, respectively. Coordinates were retrieved from the Protein Data Bank under accession code 1K3B.
PROCESSING AND ACTIVATION OF CATHEPSIN C
As mentioned in the previous section, cathepsin C is synthesized as an inactive single-chain precursor of
Procathepsin C is transported from the endoplasmic reticulum to lysosomes via the mannose-6-phosphate pathway[33,34]. Unlike other cysteine cathepsins, procathepsin C is incapable of autocatalytic processing and must be activated by other lysosomal peptidases. Cathepsins L and S were identified as the first cathepsin C activating peptidases[28]. The maturation of procathepsin C is a multistep process that involves the removal of the internal propeptide segment and the cleavage of the catalytic domain into a heavy chain and a light chain. Full processing is required to obtain the final active conformation[28]. However, it has been shown that cathepsins L and S are not required for the activation of procathepsin C in mice[35]. Similarly, a study on human neutrophil progenitor cell lines PLB-985 and HL60 revealed that complete inhibition of CatS was not sufficient to completely block the activation of procathepsin C, suggesting that other peptidases are involved in this process[36]. Recently, cathepsins F, K, and V were shown to activate procathepsin C in vitro via the same intermediate species as cathepsins L and S[29]. These results suggest that the maturation of procathepsin C is a redundant process that can be carried out in vivo by different peptidases in tissue-specific patterns.
The purpose of the tetrameric structure of cathepsin C, in contrast to the monomeric forms of other related peptidases, has remained largely unexplained. Olsen et al. suggested that the tetrameric structure merely stabilizes the interaction of the exclusion domain with the peptidase domain, thus maintaining the dipeptidyl-peptidase activity[37]. An early report also indicated that cathepsin C is an allosteric enzyme that exhibits pH- and cofactor-dependent cooperative effects[23]. However, these results were later disputed by others[14]. A partial inhibitor of cathepsin C, i.e., one that enables the enzyme to retain part of its activity in the enzyme/inhibitor complex, has been described that could presumably act via allosteric mechanisms at the level of individual subunits[31], a definitive answer to the question of cooperativity between subunits in cathepsin C is still pending.
The non-covalent interactions of the exclusion domain with the peptidase domain and between subunits are strong, and the tetramer does not dissociate in the presence of 2 M guanidinium chloride, but with further increased concentration, the oligomer is completely destroyed[38]. The recombinant peptidase domain alone has been shown to have similar endoproteolytic activity to other papain-like peptidases[39]. In addition, canine and rabbit cathepsins C have been shown to cleave substrates typical of PLP endopeptidases under certain conditions[40,41], suggesting that the exclusion domain can be removed.
PHYSIOLOGICAL AND PATHOLOGICAL FUNCTIONS OF CATHEPSIN C
As pointed out in the introduction, a major physiological role of cathepsin C is the activation of latent zymogens of effector serine peptidases in immune cells. These include four neutrophil serine peptidases (NSPs),i.e., neutrophil elastase, proteinase 3, cathepsin G[42], and the recently discovered neutrophil serine protease 4[43], as well as granzymes in cytotoxic T lymphocytes, and chymases in mast cells. All belong to the chymotrypsin-like family, classified as clan PA, family S1, subfamily S1A in the MEROPS database of peptidases[2]. All are synthesized as preproenzymes and processed to inactive zymogens upon completion of synthesis and ER import. The zymogens contain an N-terminal propeptide of two amino acid residues and are converted to their active forms via a single cleavage by cathepsin C which removes the propeptide[44-46]. This causes a conformational change that results in the formation of a catalytically competent active site. The underlying molecular mechanism is well conserved in chymotrypsin-like peptidases[47] and was also studied at the molecular level for the activation of prochymase[48].
NSPs are synthesized at the promyelocyte stage. Proper timing of the N-terminal processing is essential for their activity and optimal storage in azurophil granules. Activation normally occurs after the sorting of zymogens to pregranule/granule compartments[49]. Garwicz et al. have shown that while the N-terminal propeptide is not strictly necessary for the sorting of neutrophil cathepsin G into the granules, premature activation of the zymogen is deleterious[50]. Similarly, abnormal processing in the absence of DPPI has been shown for granzyme A, resulting in products cleaved at alternative locations by unknown peptidases[46]. NSPs can be either stored in an active form in the granules or secreted from the cell as zymogens in varying proportions, depending on the individual peptidase[51]. Activated neutrophils secrete stored, active NSPs as soluble molecules or bound to chromatin in the form of neutrophil extracellular traps[52]. Once released into the extracellular environment, these pro-inflammatory peptidases can degrade various extracellular matrix components, resulting in tissue damage and chronic inflammation[53].
Some tissue studies have provided evidence that mast cells are the predominant cathepsin C-expressing cells in the non-inflamed airways of dogs[40] and mouse skin[54]. Studies in cultured cells suggested that some cathepsin C is packaged and secreted in serine protein-rich mature granules[55]. Whether cathepsin C, which is released into the extracellular environment rich in cystatins and other potential inhibitors, can also act outside the cell by cleaving extracellular proteins remains unclear, although some studies suggest it is capable of doing so[40,41]. The repertoire of mast-cell peptidases activated by cathepsin C includes cathepsin G and chymase[56-58]. Mast cells from cathepsin C-deficient mice express normal amounts of chymase, but it has the form of an unprocessed proenzyme[45]. The involvement of cathepsin C in the activation of tryptases is less clear[45,59,60]. Recent studies suggest that cathepsin C is sufficient, but not necessary, for the complete maturation of β-tryptases in human mast cells, and that one or more other cysteine cathepsins may take over this role[60]. Experiments in animal models support a role for cathepsin C in tryptase activation, as tryptase activity is reduced but not absent in cathepsin C-deficient mice[45]. In addition, studies in animal models indicate that cathepsin C in mast cells increases the likelihood of fatal sepsis, suggesting a role for cathepsin C in regulating interleukin-6 levels by regulating the production of tryptase and other interleukin-6-degrading peptidases[61]. Similarly, granzymes in cytotoxic T lymphocytes contain N-terminal pro-dipeptides (usually Gly-Glu or Glu-Glu)[62,63]. In these cells, cathepsin C is present in secretory granules[64], and several studies have shown that cathepsin C can activate these peptidases in vitro[65-67].
Roles in immunity other than activation of serine peptidases have also been described for cathepsin C. Evidence points towards its regulatory role in macrophage polarization into the M1 phenotype via an interplay with tumor necrosis factor α (TNFα), focal adhesion kinase (FAK) and the p38/mitogen-activated protein kinase (MAPK) pathway[68,69]. Similarly, cathepsin C was shown to promote microglia M1 polarization in the brain[70].
The crucial role of cathepsin C in the activation of peptidases in immune cells suggests that its activity is also an important factor in the development of pathological conditions associated with excessive activity of these cells and their effector peptidases. These pathologies include various inflammatory and autoimmune diseases such as arthritis[71,72], asthma[40], abdominal aortic aneurysm[73], cystic fibrosis[36], pancreatitis[74], neuroinflammation[70,75] inflammatory lung diseases[36] including bronchiectasis[76], and indirectly chronic obstructive pulmonary disease[77]. Inhibition of cathepsin C activity in bone marrow was shown to be an effective method of silencing neutrophil serine peptidases, underscoring the importance of cathepsin C as a target for the treatment of these diseases[78], as discussed in more detail in the next section.
As expected for a ubiquitously expressed protein, physiological and pathological roles and substrates outside of the immune system have also been described for cathepsin C. The best known pathological examples are mutations in the CATC gene that cause the recessive hereditary diseases called Papillon-Lefèvre syndrome (PLS) and Haim-Munk syndrome (HMS)[8-10]. Symptoms of both diseases include early periodontitis, pathological thickening of the skin on the palms and feet, and increased susceptibility to infection, again highlighting the important role of cathepsin C in the immune system[11]. Neutrophil function is impaired but not completely abolished[79]. At the molecular level, mutations can result in truncated cathepsin C[9] or point mutations that cause loss of function[8,9]. At the cellular level, autophagy is impaired, and treatment of PLS patients with recombinant cathepsin C has been proposed as an approach to restore autophagic function[80].
Several studies pointed to the roles of cathepsin C in the systemic regulation of metabolism. The earliest study showed that cathepsin C inactivates the peptide hormone glucagon by sequential removal of dipeptides from the N-terminus[21]. Cathepsin C, in conjunction with plasma glutamate carboxypeptidase, was also shown to be involved in the extracellular processing of thyroglobulin, which is coupled with the release of the stress hormone thyroxin from the thyroid gland[81]. Furthermore, it has been shown to participate in lysosomal degradation of the digestive peptide hormone cholecystokinin together with tripeptidyl peptidase I (TPP-1)[82]. Due to their overlapping N-terminal exopeptidase activity, it was first suggested that cathepsin C may compensate for lack of TPP-1 activity and be used to alleviate the deleterious effects of TPP-1 loss-of-function mutations, which cause the lethal neurodegenerative lysosomal storage disorder classical late-infantile neuronal ceroid lipofuscinosis (CLN2)[82,83]. Unfortunately, studies using mouse models of the disease have invalidated this approach, as cathepsin C did not functionally compensate for the loss of TPP-I activity in the brain[84].
There is also increasing evidence that cathepsin C contributes to the progression of various cancers such as squamous cell carcinoma[54], hepatocellular carcinoma[85], gastric cancer[86], colorectal cancer[87], breast cancer[88], and so on. In these cancers, pathological cathepsin C activity originates either from tumor stromal cells[54] or directly from tumor cells[85-88], and contributes to tumorigenesis via diverse cellular and molecular mechanisms. In squamous cell carcinoma, increased activity of cathepsin C was observed in fibroblasts and bone marrow-derived cells of the tumor stroma, which promoted carcinogenesis[54]. In hepatocellular carcinoma, cathepsin C/TNFα/p38 MAPK interplay was observed similar to that contributing to macrophage M1 polarization[85], whereas in gastric and colorectal cancer, cathepsin C was associated with dysregulation of autophagy[86,87]. In breast cancer, cathepsin C was found to be secreted from tumor cells and to promote neutrophil recruitment during lung metastasis[88]. A detailed expert review on the indirect and direct roles of cathepsin C in cancer has been published recently[89].
In vitro, cathepsin C also plays a critical role in the permeabilization of the lysosomal membrane triggered by the lysosomotropic detergent L-leucyl-leucine methyl ester (LLOMe), resulting in the release of lysosomal hydrolases into the cytosol that ultimately triggers caspase-dependent apoptotic cell death. Cathepsin C has been proposed to mediate lysosomal membrane permeabilization by catalyzing the polymerization of LLOMe accumulated in lysosomes into its membranolytic polymeric form[90].
In vivo, cathepsin C activity is regulated by endogenous proteinaceous inhibitors. Like other PLP, it is amendable to inhibition by inhibitors from the cystatin family[91,92]. Of note, it is also inhibited by cystatin F[93], which is expressed only in immune cells[94]. Interestingly, inhibition depends on the proteolytic processing of cystatin F to remove its N-terminal part, which cannot be accommodated into the active site of cathepsin C[93]. Recently, it was found that Glu-Lys and Gly-Glu dipeptides derived from the pro-region of granzymes A and B, respectively, inhibit the transferase activity of cathepsin C with IC50 values < 20 mM at pH 7.4 and peptidase activity at pH 5.5 with Ki values of 20 mM and 2.5 mM for Glu-Lys and Gly-Glu, respectively[95]. Although this finding is unlikely to be considered as a strategy for pharmacological targeting of cathepsin C, it is one of the few documented examples of product inhibition in this family of peptidases and provides additional insight into the diversity of cathepsin C regulation in vivo.
PHARMACOLOGICAL INHIBITION OF CATHEPSIN C
Because of its critical roles in the activation of neutrophil and other serine peptidases, cathepsin C is considered a promising target for the treatment of diseases associated with overactivity of cells producing and secreting these peptidases, such as chronic inflammatory diseases, autoimmune diseases, and so on. Cathepsin C is by no means an easy target in these diseases, as successful anti-cathepsin C therapies must achieve a sustained inhibitory effect directly in the bone marrow to prevent the maturation of NSPs and other serine peptidase zymogens in maturing immune cells[78,96]. Cathepsin C is also an emerging target for the treatment of various cancers[89]. An expert review dedicated specifically to the pharmacological targeting of cathepsin C has been published recently[6]. Here, we shall briefly summarize the current state-of-the-art in the inhibition of cathepsin C.
Most cathepsin C inhibitors are peptide-based and form covalent bonds to the catalytic residue Cys234 using reactive diazomethyl-ketone, nitrile, semicarbazide, or vinyl-sulfone groups (i.e., “warheads”). Some of the most advanced cathepsin C inhibitors to date are collected in Table 1. One of the earliest reported cathepsin C inhibitors was Gly-Phe-CHN2[97], a substrate analog from a series of diazomethyl-ketone warhead-containing irreversible inhibitors (inactivators) of cysteine peptidases[98]. The high reactivity of the diazomethyl ketone group and instability in acidic media limited the use of such probes beyond in vitro experiments, as they are metabolically unstable and sometimes have deleterious side effects, while increased metabolic stability of such inhibitors was often accompanied by decreased inhibitory activity[99].
Selected synthetic inhibitors of human cathepsin C
| Identifier | Structure | Warhead / Type of inhibition | Potency |
| AZD5248 |  | Nitrile/reversible | pIC50 = 9.1 ± 0.1 (in vitro) pIC50 = 8.1 ± 0.1 (cell-based assay) |
| AZD7986 |  | Nitrile/reversible | pIC50 = 8.4 ± 0.17 (in vitro) pIC50 = 6.84 ± 0.33 (cell-based assay) |
| IcatC |  | Nitrile/reversible | IC50 = 15 ± 1 nM |
| GSK2793660 |  | α,β-unsaturated amide/irreversible | IC50 = 0.43-1 nM |
| Compound 54 in ref.[115] |  | Pyridine/reversible | IC50 = 57.4 ± 0.7 nM |
| Compound 11 in ref.[117] |  | Epoxide/irreversible | kinact = (5.6 ± 1.7) × 104 M-1 s-1 |
| Compound 8 in ref.[121] |  | Semicarbazide/reversible | IC50 = 31 ± 3 nM Ki = 45 ± 2 nM |
| Compound 3c in ref.[122] |  | Phosphonate (non-covalent)/reversible | Ki = 23 ± 12 nM |
| Compound 4a in ref.[122] |  | Phosphonate (non-covalent)/reversible | Ki = 51 ± 16 nM |
| Canertinib |  | Acrylamide/irreversible | IC50 = 0.12 μM |
| Compound 22 in ref.[125] |  | Acrylamide/irreversible | IC50 = 17.3 ± 0.9 nM (in vitro) IC50 = 1-5 nM (cell-based assay) |
The first cathepsin C inhibitor selected as a candidate for in vivo studies was (S)-4-amino-N-(1-cyano-2-(4'-cyano-[1,1'-biphenyl]-4-yl)ethyl)tetrahydro-2H-pyran-4-carboxamide, a reversible inhibitor with a nitrile warhead developed by AstraZeneca (UK) and later named AZD5248[100] [Table 1]. The compound had pIC50 values of 9.1 ±0.1 for purified cathepsin C in vitro and 8.1 ± 0.1 in a cell-based assay[100]. The crystal structure of AZD5248 bound into the active site of cathepsin C showed that it interacts with the non-primed sites of the enzyme as well as the N-terminal part of the exclusion domain [Figure 2][100]. It effectively inhibited the activation of NSPs in rats[101] but showed aortic binding in quantitative whole-body autoradiography studies, which could lead to potential cardiovascular toxicity. At the molecular level, aortic binding has been associated with cross-reactivity of AZD5248 with aldehydes, resulting in cross-linking with elastin in the aortic wall[102]. Therefore, novel inhibitors based on AZD5248 were prepared to overcome aortic binding, including the most advanced inhibitor to date, AZD7986 ((S)-N-((S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydrobenzo-[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide)[103]. Like AZD5248, AZD7986 is also a highly potent, reversible, and selective inhibitor of cathepsin C with pIC50 values of 8.4 for cathepsin C in vitro and cell-based assays[103]. It almost completely inhibited the activation of NSPs in primary CD34+ neutrophil progenitor cells from human bone marrow with a pIC50 value of 6.84 ± 0.33 and had favorable predicted plasma stability and clearance rate[103].
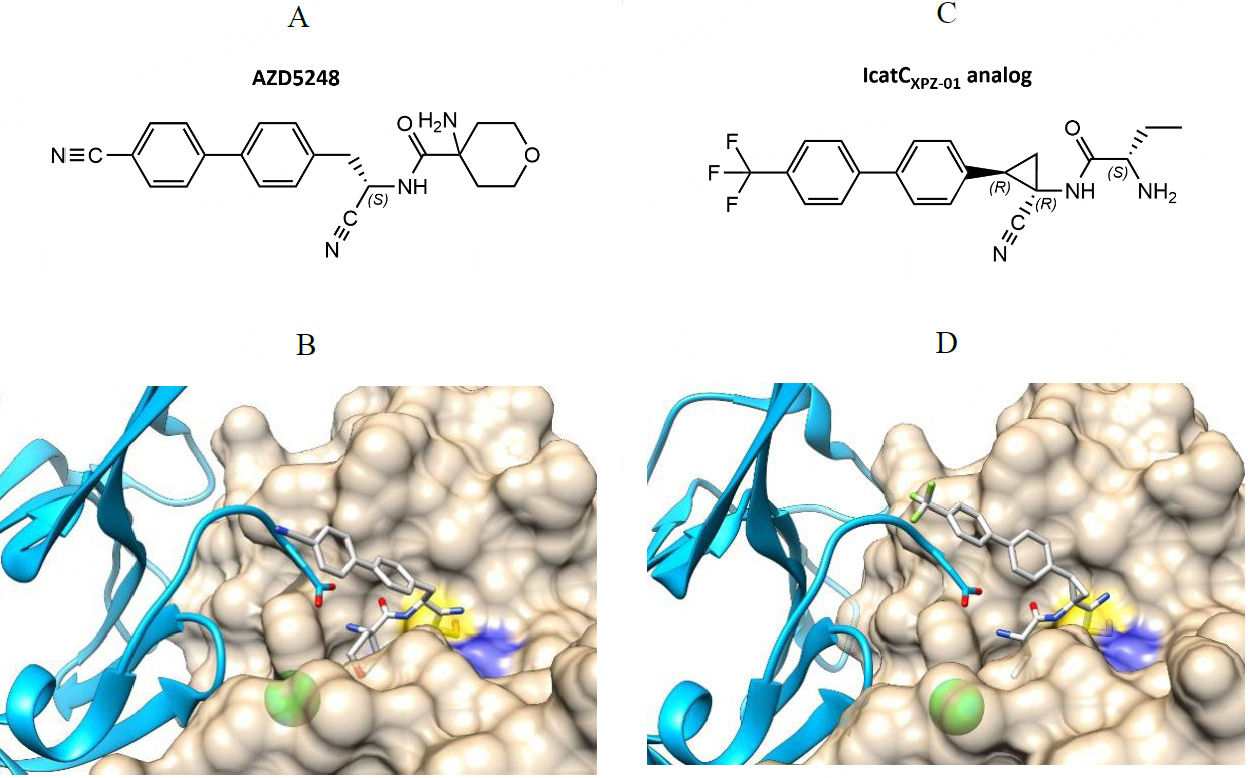
Figure 2. Reversible nitrile warhead-containing inhibitors of cathepsin C. (A) Chemical structure of compounds AZD5258 developed by AstraZeneca; (B) Crystal structure of AZDB5248 bound into the active site of cathepsin C. (PDB accession code 4CDE); (C) Chemical structure of the IcatCXPZ-01 analog used for crystallization; (D) Crystal structure of IcatCXPZ-01 analog bound into the active site of cathepsin C (PDB accession code 6IC6). Chemical structures were drawn with ChemDraw software. The crystal structures in panels B and D were drawn with UCSF Chimera software[113].
AZD7986 was the first nitrile-based cathepsin C inhibitor to reach clinical trials[104]. In 2016, Insmed, Inc (USA) announced a license agreement with AstraZeneca for exclusive worldwide rights to AZD7986. The compound was renamed INS1007 (trade name brensocatib) and was used in a trial study for the treatment of non-cystic fibrosis bronchiectasis, a persistent dilatation of the airways associated with neutrophil-mediated inflammation[76]. In the 24-week phase 2 study, which ended in 2020, patients treated with brensocatib showed reduced NSP activity, which was associated with the clinical effect of bronchiectasis[105]. These results confirmed the efficacy of brensocatib in the treatment of chronic neutrophil-related inflammatory diseases in the lung. A phase 3 clinical trial is currently underway[7]. Neutrophils and their peptidases are also associated with the severity of COVID-19[106,107]. Therefore, brensocatib has also been evaluated for its beneficial effects in hospitalized patients with COVID-19. Unfortunately, brensocatib did not improve the clinical condition of COVID-19 patients[108].
Another promising nitrile-based inhibitor of cathepsin C is the compound IcatCXPZ-01 ((S)-2-amino-N-((1R,2R)-1-cyano-2-(4’-(4-methylpiperazin-1-ylsulfonyl)biphenyl-4-yl)cyclopropyl)butanamide) [Table 1]. IcatCXPZ-01 is a reversible, potent, and cell-permeable inhibitor with an IC50 value of 15 ± 1 nM and excellent selectivity for cathepsin C[78]. The crystal structure of one of its derivatives bound to the active site of cathepsin C revealed a binding pose analogous to that of AZD5248 [Figure 2][109]. The compound successfully inhibited the activation of NSPs in bone marrow in both cell-based assays and primate experiments[78]. In mice, sufficient concentrations of IcatCXPZ-01 were accumulated in the bone marrow to inhibit cathepsin C, and subcutaneous administration of the inhibitor showed significant anti-arthritic activity in an anti-collagen-induced rheumatoid arthritis mouse model[109]. Moreover, preoperative treatment of mice prior to lung transplantation improved early graft function and decreased active NSP levels in the graft, indicating a novel potential use of cathepsin C inhibitors[110]. Despite its promising potential, there is currently no information on its further exploration as a candidate for human clinical trials.
In addition to AZD7986, two other cathepsin C inhibitors have entered clinical trials. Compound GSK2793660 [Table 1] was developed by GlaxoSmithKline (UK) and is a dipeptide-based irreversible inhibitor with an α,β-unsaturated amide-reactive group. Unfortunately, clinical trials were discontinued after several volunteers in a phase 1 study showed symptoms of epidermal desquamation of the palms and soles after repeated administration of the inhibitor, which bore some resemblance to PLS patients. In addition, no significant reduction in NSP activity was observed[111]. The third compound, BI1291583 (undisclosed structure), developed by Boehringer Ingelheim (Germany), has just started a phase 2 trial for the treatment of bronchiectasis (NCT05238675) after successfully completing phase 1[112] and is expected to be completed in Q1 of 2024.
Based on the successes and failures of these inhibitors, novel non-peptidic, non-covalent inhibitors of cathepsin C are emerging in recent years[114]. Chen et al. synthesized and characterized a series of inhibitors based on a pyridine scaffold. The best compound had a reported IC50 value of 57.4 ± 0.7 nM and was selective for cathepsin C in vitro[115] [Table 1]. Its administration also reduced NSP activation in rat bone marrow and showed anti-inflammatory activity in a rat model of COPD[115]. The compound was recently further optimized to improve its pharmacokinetic properties[116]. Further research will shed light on the efficacy and safety of these compounds.
In addition, a substantial number of cathepsin C inhibitors have been reported that have provided only in vitro data and no in vivo follow-up studies to date. Using a structure-based approach, Radzey et al. used E-64c hydrazide as a lead structure for the development of irreversible cathepsin C inhibitors[117]. The best resulting inhibitor, (2S,3S)-3-(2-butylhydrazine carbonyl)-N-((S)-1-(isopentylamino)-4-methyl-1-oxopentan-2-yl)oxirane-2-carboxamide [Table 1], exhibits significantly improved potency (kinact = (5.6 ± 0.17) × 104 M-1 s-1) compared to E-64c-hydrazide (kinact = 140 ± 5 M-1 s-1). It also reacts more rapidly with cathepsin C than with cathepsin L, which is the opposite of E-64c and its hydrazide, which have a strong preference for cathepsin L[117]. The authors therefore proposed this compound as a starting point for the development of optimized inhibitors that bind to the S1'-S2' sites of cathepsin C[117].
Azapeptides, i.e., peptide analogs in which one or more amino acids have been replaced by a semicarbazide group, have been reported as inhibitors of hepatitis C virus NS3 peptidase[118], human rhinovirus 3C peptidase[119], and several papain-like cysteine peptidases[120]. Azapeptides with weak leaving groups cannot acylate the enzyme and therefore show competitive inhibition, whereas azapeptides with reactive leaving groups form carbamoyl-enzyme complexes that are more stable than normal acyl enzymes. For cathepsin C, one of the best inhibitors from this class was 1-(2S-2-aminobutanoyl)-4-{2S-N-[2S-3-(m-fluoro- phenyl)propan-2-yl-amide]-4-phenylbutan-2-yl-amide}semi- carbazide [Table 1] with an IC50 value of 31 ± 3 nM and a Ki value of 45 ± 2 nM. The compound acted as a reversible, competitive inhibitor and was selective for cathepsin C. It was not cytotoxic to HepG2 cells and showed about 50% inhibition of intracellular cathepsin C activity in this cell line[121].
Drag et al. described a series of phosphonate dipeptide analogs as non-covalent inhibitors of
Moreover, two known epidermal growth factor receptor (EGFR) inhibitors containing Cys-reactive acrylamide warheads were identified as cathepsin C inhibitors by activity-based protein profiling, highlighting the potential of drug repurposing for the targeting of cathepsin C. In 2016, Canertinib, a former drug candidate developed by Pfizer (USA) for the treatment of cancer, was identified as a potent
Naturally occurring compounds are a rich source of pharmacologically active molecules and inhibition of cathepsin C by such compounds has also been investigated to some extent. A recent study has shown that polyglucoside from the vine Triperygium wilfordii inhibited cathepsin C activity in the serum, synovial fluid, and tissues in a rat model of collagen-induced arthritis, highlighting the possibility that the suppression of rheumatoid arthritis traditionally associated with Tripergyium wilfordii polyglucoside may be related to the inhibition of cathepsin C and downstream serine peptidases[126]. In addition, we found that caffeic acid and its derivatives inhibit cysteine cathepsins, including cathepsin C[31,127], in vitro. Since polyphenolic compounds such as caffeic acid have a broad spectrum of pharmacological activity and biological targets, their use for specific targeting of individual protein targets is not feasible. Nevertheless, caffeic acid is considered a promising pharmacophore for the development of specific inhibitors[128]. Alternative methods for targeting cathepsin C are also emerging. We have identified 3’-nitrophthalanilic acid as a partial inhibitor of cathepsins K and C that presumably acts by binding outside of the active site[31,129]. Such allosteric drugs are a popular and promising approach for the targeting of membrane receptors[130] and strategies of targeting sites away from the active site have also been explored in several PLPs, including human cathepsin K[131,132] and plasmodial cathepsin C homologs[133]. However, in the case of human cathepsin C, the development of specific partial inhibitors suitable for use in cell culture and in vivo and the evaluation of the efficacy of such an approach are still in their infancy.
CONCLUSION
Although cathepsin C was among the first peptidases to be identified, interest in this peptidase as a drug target has only emerged in the last two decades, primarily leading to facilitated investigation of its physiological and pathological roles. With two inhibitors currently in clinical trials for the treatment of non-cystic fibrosis bronchiectasis, cathepsin C is currently the most important and promising human cysteine cathepsin from a clinical perspective. Considering the recent failures in the field of peptidase targeting, e.g., the cathepsin K inhibitor odanacatib (Merck & Co., USA) for the treatment of osteoporosis which was discontinued after the completion of phase 3 clinical trials[134], the outcomes of these trials may be crucial to (re)establish not only cathepsin C, but cysteine cathepsins and peptidases in general as valid drug targets in human diseases. From the perspective of basic protein science, cathepsin C is proving to be an interesting model protein for studying the molecular mechanisms and evolution of protein oligomerization[4]. Further research in this direction could, in turn, provide novel strategies for the targeting of this important peptidase.
DECLARATIONS
Authors’ contributionsWrote and revised the manuscript: Stojkovska Docevska M, Novinec M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe authors acknowledge the financial support of the Slovenian Research Agency (project grant No. N1-0211 to MN and research core funding No. P1-0179).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Tallan HH, Jones ME, Fruton JS. On the proteolytic enzymes of animal tissues. X. beef spleen cathepsin C. J Biol Chem 1952;194:793-805.
2. Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 2018;46:D624-32.
3. Novinec M, Lenarčič B. Papain-like peptidases: structure, function, and evolution. Biomol Concepts 2013;4:287-308.
5. Wu C, Jin X, Tsueng G, Afrasiabi C, Su AI. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res 2016;44:D313-6.
6. Korkmaz B, Caughey GH, Chapple I, et al. Therapeutic targeting of cathepsin C: from pathophysiology to treatment. Pharmacol Ther 2018;190:202-36.
7. Chalmers JD, Usansky H, Rubino CM, et al. Pharmacokinetic/pharmacodynamic evaluation of the dipeptidyl peptidase 1 inhibitor brensocatib for non-cystic fibrosis bronchiectasis. Clin Pharmacokinet 2022;61:1457-69.
8. Nakano A, Nomura K, Nakano H, et al. Papillon-Lefèvre syndrome: mutations and polymorphisms in the cathepsin C gene. J Invest Dermatol 2001;116:339-43.
9. Hart TC, Hart PS, Bowden DW, et al. Mutations of the cathepsin C gene are responsible for Papillon-Lefèvre syndrome. J Med Genet 1999;36:881-7.
10. Hart TC, Hart PS, Michalec MD, et al. Haim-munk syndrome and papillon-Lefèvre syndrome are allelic mutations in cathepsin C. J Med Genet 2000;37:88-94.
11. Sreeramulu B, Shyam ND, Ajay P, Suman P. Papillon-Lefèvre syndrome: clinical presentation and management options. Clin Cosmet Investig Dent 2015;7:75-81.
12. Turk V, Stoka V, Vasiljeva O, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012;1824:68-88.
13. Turk D, Janjic V, Stern I, et al. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J 2001;20:6570-82.
14. Cigic B, Pain RH. Location of the binding site for chloride ion activation of cathepsin C. Eur J Biochem 1999;264:944-51.
15. Lindley H. The specificity of dipeptidyl aminopeptidase I (cathepsin C) and its use in peptide sequence studies. Biochem J 1972;126:683-5.
16. Wang F, Krai P, Deu E, et al. Biochemical characterization of plasmodium falciparum dipeptidyl aminopeptidase 1. Mol Biochem Parasitol 2011;175:10-20.
17. de Vries LE, Sanchez MI, Groborz K, et al. Characterization of P. falciparum dipeptidyl aminopeptidase 3 specificity identifies differences in amino acid preferences between peptide-based substrates and covalent inhibitors. FEBS J 2019;286:3998-4023.
18. Poreba M, Mihelic M, Krai P, et al. Unnatural amino acids increase activity and specificity of synthetic substrates for human and malarial cathepsin C. Amino Acids 2014;46:931-43.
19. Łęgowska M, Hamon Y, Wojtysiak A, et al. Development of the first internally-quenched fluorescent substrates of human cathepsin C: the application in the enzyme detection in biological samples. Arch Biochem Biophys 2016;612:91-102.
20. Metrione RM, MacGeorge NL. The mechanism of action of dipeptidyl aminopeptidase. Inhibition by amino acid derivatives and amines; activation by aromatic compounds. Biochemistry 1975;14:5249-52.
21. Mcdonald JK, Callahan PX, Zeitman BB, Ellis S. Inactivation and degradation of glucagon by dipeptidyl aminopeptidase I (Cathepsin C) of rat liver. J Biol Chem 1969;244:6199-208.
22. Dolenc I, Turk B, Pungercic G, Ritonja A, Turk V. Oligomeric structure and substrate induced inhibition of human cathepsin C. J Biol Chem 1995;270:21626-31.
24. Nilsson KK, Fruton JS. Polymerization reactions catalyzed by intracellular proteinases. iv. factors influencing the polymerization of dipeptide amides by cathepsin C. Biochemistry 1964;3:1220-4.
25. Gittel C, Schmidtchen FP. Directed N-terminal elongation of unprotected peptides catalyzed by cathepsin c in water. Bioconjug Chem 1995;6:70-6.
26. Tavano OL. Protein hydrolysis using proteases: an important tool for food biotechnology. J Mol Catal B-Enzym 2013;90:1-11.
27. Cigić B, Dahl SW, Pain RH. The residual pro-part of cathepsin C fulfills the criteria required for an intramolecular chaperone in folding and stabilizing the human proenzyme. Biochemistry 2000;39:12382-90.
28. Dahl SW, Halkier T, Lauritzen C, et al. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 2001;40:1671-8.
29. Lamort AS, Hamon Y, Czaplewski C, et al. Processing and maturation of cathepsin C zymogen: a biochemical and molecular modeling analysis. Int J Mol Sci 2019;20:4747.
30. Santilman V, Jadot M, Mainferme F. Importance of the propeptide in the biosynthetic maturation of rat cathepsin C. Eur J Cell Biol 2002;81:654-63.
31. Rebernik M, Snoj T, Klemenčič M, Novinec M. Interplay between tetrameric structure, enzymatic activity and allosteric regulation of human dipeptidyl-peptidase I. Arch Biochem Biophys 2019;675:108121.
32. Lauritzen C, Pedersen J, Madsen MT, Justesen J, Martensen PM, Dahl SW. Active recombinant rat dipeptidyl aminopeptidase I (cathepsin C) produced using the baculovirus expression system. Protein Expr Purif 1998;14:434-42.
33. Burge V, Mainferme F, Wattiaux R. Transient membrane association of the precursors of cathepsin C during their transfer into lysosomes. Biochem J 1991;275:797-800.
34. Muno D, Ishidoh K, Ueno T, Kominami E. Processing and transport of the precursor of cathepsin C during its transfer into lysosomes. Arch Biochem Biophys 1993;306:103-10.
35. Clair J, Shi GP, Sutherland RE, Chapman HA, Caughey GH, Wolters PJ. Cathepsins L and S are not required for activation of dipeptidyl peptidase I (cathepsin C) in mice. Biol Chem 2006;387:1143-6.
36. Hamon Y, Legowska M, Hervé V, et al. Neutrophilic cathepsin C is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J Biol Chem 2016;291:8486-99.
37. Olsen JG, Kadziola A, Lauritzen C, Pedersen J, Larsen S, Dahl SW. Tetrameric dipeptidyl peptidase I directs substrate specificity by use of the residual pro-part domain. FEBS Lett 2001;506:201-6.
38. Cigić B, Pain RH. Competitive inhibition of cathepsin C by guanidinium ions and reexamination of substrate inhibition. Biochem Biophys Res Commun 1999;258:6-10.
39. Rebernik M, Lenarčič B, Novinec M. The catalytic domain of cathepsin C (dipeptidyl-peptidase I) alone is a fully functional endoprotease. Protein Expr Purif 2019;157:21-7.
40. Wolters PJ, Laig-Webster M, Caughey GH. Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways. Am J Respir Cell Mol Biol 2000;22:183-90.
41. Kuribayashi M, Yamada H, Ohmori T, Yanai M, Imoto T. Endopeptidase activity of cathepsin C, dipeptidyl aminopeptidase I, from bovine spleen. J Biochem 1993;113:441-9.
42. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 2006;6:541-50.
43. Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci U S A 2012;109:6229-34.
44. Salvesen G, Enghild JJ. An unusual specificity in the activation of neutrophil serine proteinase zymogens. Biochemistry 1990;29:5304-8.
45. Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem 2001;276:18551-6.
46. Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B
47. Freer ST, Kraut J, Robertus JD, Wright HT, Xuong NH. Chymotrypsinogen: 2.5-angstrom crystal structure, comparison with alpha-chymotrypsin, and implications for zymogen activation. Biochemistry 1970;9:1997-2009.
48. Reiling KK, Krucinski J, Miercke LJ, Raymond WW, Caughey GH, Stroud RM. Structure of human pro-chymase: a model for the activating transition of granule-associated proteases. Biochemistry 2003;42:2616-24.
49. Gullberg U, Andersson E, Garwicz D, Lindmark A, Olsson I. Biosynthesis, processing and sorting of neutrophil proteins: insight into neutrophil granule development. Eur J Haematol 1997;58:137-53.
50. Garwicz D, Lindmark A, Persson A, Gullberg U. On the role of the proform-conformation for processing and intracellular sorting of human cathepsin G. Blood 1998;92:1415-22.
51. Garwicz D, Lennartsson A, Jacobsen SE, Gullberg U, Lindmark A. Biosynthetic profiles of neutrophil serine proteases in a human bone marrow-derived cellular myeloid differentiation model. Haematologica 2005;90:38-44.
52. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532-5.
53. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726-59.
54. Ruffell B, Affara NI, Cottone L, et al. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev 2013;27:2086-98.
55. Wolters PJ, Raymond WW, Blount JL, Caughey GH. Regulated expression, processing, and secretion of dog mast cell dipeptidyl peptidase I. J Biol Chem 1998;273:15514-20.
56. Dikov MM, Springman EB, Yeola S, Serafin WE. Processing of procarboxypeptidase A and other zymogens in murine mast cells. J Biol Chem 1994;269:25897-904.
57. Gullberg U, Lindmark A, Nilsson E, Persson A, Olsson I. Processing of human cathepsin G after transfection to the rat basophilic/mast cell tumor line RBL. J Biol Chem 1994;269:25219-25.
58. McEuen AR, Ashworth DM, Walls AF. The conversion of recombinant human mast cell prochymase to enzymatically active chymase by dipeptidyl peptidase I is inhibited by heparin and histamine. Eur J Biochem 1998;253:300-8.
59. Le QT, Min HK, Xia HZ, Fukuoka Y, Katunuma N, Schwartz LB. Promiscuous processing of human alphabeta-protryptases by cathepsins L, B, and C. J Immunol 2011;186:7136-43.
60. Le QT, Gomez G, Zhao W, et al. Processing of human protryptase in mast cells involves cathepsins L, B, and C. J Immunol 2011;187:1912-8.
61. Clair J, Pham CT, Villalta SA, Caughey GH, Wolters PJ. Mast cell dipeptidyl peptidase I mediates survival from sepsis. J Clin Invest 2004;113:628-34.
62. Jenne D, Rey C, Haefliger JA, Qiao BY, Groscurth P, Tschopp J. Identification and sequencing of cDNA clones encoding the granule-associated serine proteases granzymes D, E, and F of cytolytic T lymphocytes. Proc Natl Acad Sci U S A 1988;85:4814-8.
63. Caputo A, Garner R, Winkler U, Hudig D, Bleackley R. Activation of recombinant murine cytotoxic cell proteinase-1 requires deletion of an amino-terminal dipeptide. J Biol Chem 1993;268:17672-5.
64. Brown GR, McGuire MJ, Thiele DL. Dipeptidyl peptidase I is enriched in granules of
65. Mcguire M, Lipsky P, Thiele D. Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem 1993;268:2458-67.
66. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004;104:2840-8.
67. Pham CT, Thomas DA, Mercer JD, Ley TJ. Production of fully active recombinant murine granzyme B in yeast. J Biol Chem 1998;273:1629-33.
68. Alam S, Liu Q, Liu S, et al. Up-regulated cathepsin C induces macrophage M1 polarization through FAK-triggered p38 MAPK/NF-κB pathway. Exp Cell Res 2019;382:111472.
69. Dai J, Liu J, Zhang Q, et al. Cathepsin C is involved in macrophage M1 polarization via p38/MAPK pathway in sudden cardiac death. Cardiovasc Ther 2021;2021:6139732.
70. Liu Q, Zhang Y, Liu S, et al. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca2+-dependent PKC/p38MAPK/NF-κB pathway. J Neuroinflamm 2019;16:1-18.
71. Adkison AM, Kelley DG, Pham CTN, et al. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis find the latest version: dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development. J Clin Invest 2002;109:363-71.
72. Chu Y, Guo Y, Walls AF, Zhou X. The regulatory role of dipeptidyl peptidase I on the activation of immune granulocytes. Cell Biol Int 2017;41:1093-102.
73. Pagano MB, Bartoli MA, Ennis TL, et al. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A 2007;104:2855-60.
74. John DS, Aschenbach J, Krüger B, et al. Deficiency of cathepsin C ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of E-cadherin. J Biol Chem 2019;294:697-707.
75. Fan K, Wu X, Fan B, et al. Up-regulation of microglial cathepsin C expression and activity in lipopolysaccharide-induced neuroinflammation. J Neuroinflamm 2012;9:1-13.
76. Keir HR, Shoemark A, Dicker AJ, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med 2021;9:873-84.
77. Kim S, Nadel JA. Role of neutrophils in mucus hypersecretion in COPD and implications for therapy. Treat Respir Med 2004;3:147-59.
78. Guarino C, Hamon Y, Croix C, et al. Prolonged pharmacological inhibition of cathepsin C results in elimination of neutrophil serine proteases. Biochem Pharmacol 2017;131:52-67.
79. Sanchez Klose FP, Björnsdottir H, Dahlstrand Rudin A, et al. A rare CTSC mutation in Papillon-Lefèvre syndrome results in abolished serine protease activity and reduced NET formation but otherwise normal neutrophil function. PLoS One 2021;16:e0261724.
80. Bullón P, Castejón-Vega B, Román-Malo L, et al. Autophagic dysfunction in patients with Papillon-Lefèvre syndrome is restored by recombinant cathepsin C treatment. J Allergy Clin Immunol 2018;142:1131-1143.e7.
81. Suban D, Zajc T, Renko M, Turk B, Turk V, Dolenc I. Cathepsin C and plasma glutamate carboxypeptidase secreted from Fischer rat thyroid cells liberate thyroxin from the N-terminus of thyroglobulin. Biochimie 2012;94:719-26.
82. Bernardini F, Warburton MJ. Lysosomal degradation of cholecystokinin-(29-33)-amide in mouse brain is dependent on tripeptidyl peptidase-I: implications for the degradation and storage of peptides in classical late-infantile neuronal ceroid lipofuscinosis. Biochem J 2002;366:521-9.
83. Sleat DE, Donnelly RJ, Lackland H, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 1997;277:1802-5.
84. Kim KH, Pham CT, Sleat DE, Lobel P. Dipeptidyl-peptidase I does not functionally compensate for the loss of tripeptidyl-peptidase I in the neurodegenerative disease late-infantile neuronal ceroid lipofuscinosis. Biochem J 2008;415:225-32.
85. Zhang GP, Yue X, Li SQ. Cathepsin C interacts with TNF-α/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res Treat 2020;52:10-23.
86. Kim S, Lee S-I, Kim N, et al. Decursin inhibits cell growth and autophagic flux in gastric cancer via suppression of cathepsin C. Am J Cancer Res 2021;11:1304.
87. Khaket TP, Singh MP, Khan I, Bhardwaj M, Kang SC. Targeting of cathepsin C induces autophagic dysregulation that directs ER stress mediated cellular cytotoxicity in colorectal cancer cells. Cell Signal 2018;46:92-102.
88. Xiao Y, Cong M, Li J, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021;39:423-437.e7.
89. Korkmaz B, Lamort AS, Domain R, et al. Cathepsin C inhibition as a potential treatment strategy in cancer. Biochem Pharmacol 2021;194:114803.
90. Kavčič N, Butinar M, Sobotič B, et al. Intracellular cathepsin C levels determine sensitivity of cells to leucyl-leucine methyl ester-triggered apoptosis. FEBS J 2020;287:5148-66.
91. Nicklin MJ, Barrett AJ. Inhibition of cysteine proteinases and dipeptidyl peptidase I by egg-white cystatin. Biochem J 1984;223:245-53.
92. Tušar L, Usenik A, Turk B, Turk D. Mechanisms applied by protein inhibitors to inhibit cysteine proteases. Int J Mol Sci 2021;22:997.
93. Hamilton G, Colbert JD, Schuettelkopf AW, Watts C. Cystatin F is a cathepsin C-directed protease inhibitor regulated by proteolysis. EMBO J 2008;27:499-508.
94. Halfon S, Ford J, Foster J, et al. Leukocystatin, a new Class II cystatin expressed selectively by hematopoietic cells. J Biol Chem 1998;273:16400-8.
95. Božič J, Dolenc I. Feedback regulation of cathepsin C by the propeptide dipeptides of granzymes A and B. ACSi 2019;66:501-9.
96. Méthot N, Rubin J, Guay D, et al. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro-enzyme processing. J Biol Chem 2007;282:20836-46.
97. Mølgaard A, Arnau J, Lauritzen C, Larsen S, Petersen G, Pedersen J. The crystal structure of human dipeptidyl peptidase I (cathepsin C) in complex with the inhibitor Gly-Phe-CHN2. Biochem J 2007;401:645-50.
98. Green GD, Shaw E. Peptidyl diazomethyl ketones are specific inactivators of thiol proteinases. J Biol Chem 1981;256:1923-8.
99. Laine DI, Busch-Petersen J. Inhibitors of cathepsin C (dipeptidyl peptidase I). Expert Opin Ther Pat 2010;20:497-506.
100. Furber M, Tiden AK, Gardiner P, et al. Cathepsin C inhibitors: property optimization and identification of a clinical candidate. J Med Chem 2014;57:2357-67.
101. Gardiner P, Wikell C, Clifton S, Shearer J, Benjamin A, Peters SA. Neutrophil maturation rate determines the effects of dipeptidyl peptidase 1 inhibition on neutrophil serine protease activity. Br J Pharmacol 2016;173:2390-401.
102. Bragg RA, Brocklehurst S, Gustafsson F, et al. Aortic binding of AZD5248: mechanistic insight and reactivity assays to support lead optimzation. Chem Res Toxicol 2015;28:1991-9.
103. Doyle K, Lönn H, Käck H, et al. Discovery of second generation reversible covalent DPP1 inhibitors leading to an oxazepane amidoacetonitrile based clinical candidate (AZD7986). J Med Chem 2016;59:9457-72.
104. Palmér R, Mäenpää J, Jauhiainen A, et al. Dipeptidyl peptidase 1 inhibitor AZD7986 induces a sustained, exposure-dependent reduction in neutrophil elastase activity in healthy subjects. Clin Pharmacol Ther 2018;104:1155-64.
105. Chalmers JD, Haworth CS, Metersky ML, et al. WILLOW Investigators. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med 2020;383:2127-37.
106. Sinha S, Rosin NL, Arora R, et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat Med 2022;28:201-11.
107. Seren S, Derian L, Keleş I, et al. Proteinase release from activated neutrophils in mechanically ventilated patients with non-COVID-19 and COVID-19 pneumonia. Eur Respir J 2021;57:2003755.
108. Keir HR, Long MB, Abo-Leyah H, et al. STOP-COVID19 Investigators. Dipeptidyl peptidase-1 inhibition in patients hospitalised with COVID-19: a multicentre, double-blind, randomised, parallel-group, placebo-controlled trial. Lancet Respir Med 2022;10:1119-28.
109. Korkmaz B, Lesner A, Wysocka M, et al. Structure-based design and
110. Rehm SRT, Smirnova NF, Morrone C, et al. Premedication with a cathepsin C inhibitor alleviates early primary graft dysfunction in mouse recipients after lung transplantation. Sci Rep 2019;9:9925.
111. Miller BE, Mayer RJ, Goyal N, et al. Epithelial desquamation observed in a phase I study of an oral cathepsin C inhibitor (GSK2793660). Br J Clin Pharmacol 2017;83:2813-20.
112. Badorrek P, Diefenbach C, Kögler H, et al. Phase I characterization of the novel cathepsin C inhibitor BI 1291583. Am J Resp Crit Care 2022;205:A4777.
113. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605-12.
114. Shen XB, Chen X, Zhang ZY, Wu FF, Liu XH. Cathepsin C inhibitors as anti-inflammatory drug discovery: challenges and opportunities. Eur J Med Chem 2021;225:113818.
115. Chen X, Yan Y, Zhang Z, et al. Discovery and
116. Chen X, Yan Y, Du J, et al. Non-peptidyl non-covalent cathepsin C inhibitoEEr bearing a unique thiophene-substituted pyridine: design, structure-activity relationship and anti-inflammatory activity
117. Radzey H, Rethmeier M, Klimpel D, Grundhuber M, Sommerhoff CP, Schaschke N. E-64c-hydrazide: a lead structure for the development of irreversible cathepsin C inhibitors. ChemMedChem 2013;8:1314-21.
118. Zhang R, Durkin JP, Windsor WT. Azapeptides as inhibitors of the hepatitis C virus NS3 serine protease. Bioorg Med Chem Lett 2002;12:1005-8.
119. Venkatraman S, Kong J, Nimkar S, Wang QM, Aubé J, Hanzlik RP. Design, synthesis, and evaluation of azapeptides as substrates and inhibitors for human rhinovirus 3C protease. Bioorg Med Chem Lett 1999;9:577-80.
120. Verhelst SH, Witte MD, Arastu-Kapur S, Fonovic M, Bogyo M. Novel aza peptide inhibitors and active-site probes of papain-family cysteine proteases. Chembiochem 2006;7:943-50.
121. Bondebjerg J, Fuglsang H, Valeur KR, et al. Novel semicarbazide-derived inhibitors of human dipeptidyl peptidase I (hDPPI). Bioorg Med Chem 2005;13:4408-24.
122. Drąg M, Wieczerzak E, Pawełczak M, Berlicki Ł, Grzonka Z, Kafarski P. Toward very potent, non-covalent organophosphonate inhibitors of cathepsin C and related enzymes by 2-amino-1-hydroxy-alkanephosphonates dipeptides. Biochimie 2013;95:1640-9.
123. Jewgiński MP, Makowski M, Pawełczak M, et al. Synthesis of hybrid tripeptide peptidomimetics containing dehydroamino acid and aminophosphonic acid in the chain and evaluation of their activity toward cathepsin C. Chem Biodivers 2022;19:e202101019.
124. Sun H, Ren Y, Hou W, et al. Focusing on probe-modified peptides: a quick and effective method for target identification. Chem Commun 2016;52:10225-8.
125. Hou W, Sun H, Ma Y, Liu C, Zhang Z. Identification and optimization of novel cathepsin C inhibitors derived from EGFR inhibitors. J Med Chem 2019;62:5901-19.
126. Wang J, Chu Y, Zhou X. Inhibitory effect of triperygium wilfordii polyglucoside on dipeptidyl peptidase I
127. Ulčakar L, Novinec M. Inhibition of human cathepsins B and L by caffeic acid and its derivatives. Biomolecules 2020;11:31.
128. Touaibia M, Jean-François J, Doiron J. Caffeic acid, a versatile pharmacophore: an overview. Mini Rev Med Chem 2011;11:695-713.
129. Novinec M, Lenarčič B, Baici A. Probing the activity modification space of the cysteine peptidase cathepsin K with novel allosteric modifiers. PLoS One 2014;9:e106642.
130. Changeux JP, Christopoulos A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 2016;166:1084-102.
131. Novinec M, Korenč M, Caflisch A, Ranganathan R, Lenarčič B, Baici A. A novel allosteric mechanism in the cysteine peptidase cathepsin K discovered by computational methods. Nat Commun 2014;5:3287.
132. Panwar P, Søe K, Guido RV, Bueno RV, Delaisse JM, Brömme D. A novel approach to inhibit bone resorption: exosite inhibitors against cathepsin K. Br J Pharmacol 2016;173:396-410.
133. Sanchez MI, de Vries LE, Lehmann C, et al. Identification of plasmodium dipeptidyl aminopeptidase allosteric inhibitors by high throughput screening. PLoS One 2019;14:e0226270.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Stojkovska Docevska M, Novinec M. Cathepsin C: structure, function, and pharmacological targeting. Rare Dis Orphan Drugs J 2023;2:14. http://dx.doi.org/10.20517/rdodj.2023.09
AMA Style
Stojkovska Docevska M, Novinec M. Cathepsin C: structure, function, and pharmacological targeting. Rare Disease and Orphan Drugs Journal. 2023; 2(3): 14. http://dx.doi.org/10.20517/rdodj.2023.09
Chicago/Turabian Style
Stojkovska Docevska, Milena, Marko Novinec. 2023. "Cathepsin C: structure, function, and pharmacological targeting" Rare Disease and Orphan Drugs Journal. 2, no.3: 14. http://dx.doi.org/10.20517/rdodj.2023.09
ACS Style
Stojkovska Docevska, M.; Novinec M. Cathepsin C: structure, function, and pharmacological targeting. Rare. Dis. Orphan. Drugs. J. 2023, 2, 14. http://dx.doi.org/10.20517/rdodj.2023.09
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 2
likes
Like This Article 2
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.