
Pharmacovigilance of gene therapy medicinal products
 0
0 , ...
, ... Abstract
Gene therapy medicinal products (GTMPs) may generate unexpected risks to public health and individual patients. Pharmacovigilance serves the purpose of detecting, assessing, understanding, and preventing adverse effects or any other medicine-related problems. This article aims to provide an overview of the significance of pharmacovigilance and particularities in the domain of gene therapy with a brief description of the European regulatory framework. Viral vectors, insertional mutagenesis, viral latency or reactivation, as well as duration and accuracy of the patient’s clinical follow-up, are some of the main concerns. Two recent examples of signals illustrate some of these safety issues. The first concerns onasemnogene abeparvovec, an adeno-associated virus-based gene replacement therapy for treating spinal muscular atrophy. This GTMP initially raised safety concerns about hepatotoxicity, cardiotoxicity, and neurotoxicity, necessitating recommendations for close monitoring. During the post-marketing period, two fatal cases of acute liver failure led the European Medicine Agency (EMA) to strengthen the recommendations for liver function monitoring. The second example pertains to betibeglogene autotemcel, a genetically modified autologous CD34+ cell-enriched population that contains hematopoietic stem cells transduced with lentiviral vectors encoding the β A-T87Q-globin gene. After the report of a case of acute myeloid leukemia in a patient treated with an investigational product using the same lentiviral vector, the European Commission triggered a regulatory safety procedure. After careful assessment, the EMA’s ad hoc safety committees stated that the viral vector was unlikely to be the cause. However, due to commercial reasons, betibeglogene autotemcel was finally withdrawn from the market by the marketing authorization holder. Pharmacovigilance activities and systems continuously evolve to keep pace with advancements in new therapies and technologies. They must also address varied situations and adhere to evolving regulations. GTMPs likely stand out as one of the most demanding areas within pharmacovigilance. Therefore, their effective oversight requires the full commitment of pharmacological and clinical experts, as well as active involvement from patients, to ensure optimal outcomes.
Keywords
INTRODUCTION
Gene therapy can provide clinical results and outcomes that remained unbelievable until a few decades ago. However, the novelty, complexity, and technical specificity of GTMPs may generate unexpected risks to public health and individual patients[1]. These new sophisticated drugs require collaboration among regulatory bodies, marketing authorization holders (MAH), and healthcare professionals to ensure the delivery, evaluation, administration, and supervision of the safest and most effective treatments for a broad spectrum of patients in need.
Therefore, pharmacovigilance (PV) activity and risk management systems in clinical settings are essential. Since 2008, all advanced therapy medicinal products (ATMPs), including GTMPs, must be authorized in the European Union (EU) via a centralized authorization procedure (CAP).
This article aims to provide an overview of the particularities of PV in the domain of GTMPs, first with a brief description of the European regulatory framework, and second by illustrating the management of recent safety signals, especially for two GTMPs: onasemnogene abeparvovec (Zolgensma®) and betibeglogen autotemcel (Zinteglo®).
THE EUROPEAN REGULATORY FRAMEWORK FOR PHARMACOVIGILANCE
PV semantic reminder
PV is the discipline focused on detecting, assessing, understanding, and preventing adverse effects or any other medicine-related problems[2]. Its primary objective is to safeguard patient safety in the utilization of pharmaceuticals, including biologics, plasma-derived therapies, cellular and gene therapies[3]. PV is based on specific terminology and tools. An adverse drug reaction (ADR) is a response to a medicinal product that is noxious and unintended, characterized by a suspected causal relationship with the medicinal product[4]. Each country develops its PV system, coordinated by its national health regulatory authority[2,4]. In the European Union, PV systems have to be in accordance with the Good Pharmacovigilance Practices (GVPs), a quality standard for monitoring the safety of medicines[4]. These GVPs extend their application to marketing-authorization holders (MAHs) and the EMA. In other countries (USA, Canada, UK, Japan…), GVP may differ but keep the same objectives. A risk management system is a set of PV activities and interventions to identify, characterize, prevent, or minimize risks relating to a medicinal product, including the assessment of the effectiveness of these activities and interventions[4]. Safety concerns are important risks and missing information identified during clinical development, which will be closely monitored during the post-marketing phase. A risk management plan (RMP) is aimed to describe the system managing safety concerns. Direct healthcare professional communication (DHPC) is a communication intervention by a MAH or a competent authority for directly delivering important safety information on a medicinal product to individual healthcare professionals. It informs about the need to take action or adjust their practices[5]. Among all the EMA expert committees, three play pivotal roles in the oversight of ATMP and GT: the Committee for Medicinal Products for Human Use (CHMP), the Pharmacovigilance Risk Assessment Committee (PRAC), and the Committee for Advanced Therapies (CAT). The CAT holds the specific responsibility of evaluating the quality, safety, and efficacy of ATMPs, while also keeping abreast of scientific developments in this domain[6]. It furnishes its assessment of each ATMP to the CHMP, which ultimately delivers the final verdict regarding the marketing authorization. The CAT can also be consulted about any scientific issues concerning ATMPs. The PRAC will assess the risk management of ATMPs, provide recommendations to CHMP, and monitor the safety of the products throughout their lifecycle, including instances where delayed adverse reactions are anticipated or occur post-administration.
Particularities of pharmacovigilance for GTMPs
ATMPs are new, sometimes complex, and involve technical specificity, and thus may cause new risks to patients. The European guideline on safety and efficacy follow-up-risk management of ATMPs applies to GTMPs and describes rules in order to facilitate early detection of specific risks and effective mitigation of their consequences to public health or individual patients [Figure 1][7].
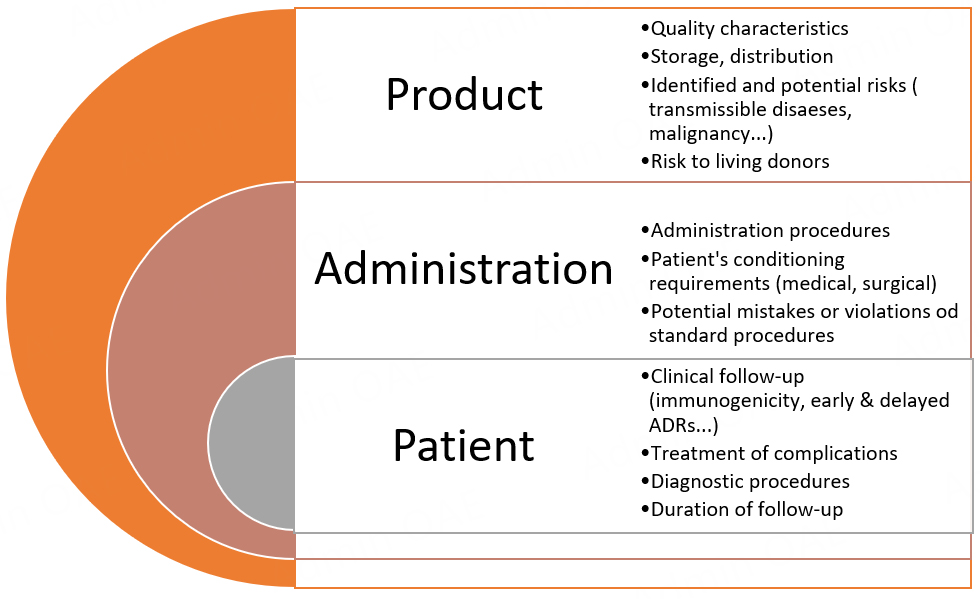
Figure 1. Summary of the main specific issues to be addressed for advanced therapy medicinal products and their application in gene therapy medicinal products[7].
For example, regarding the pharmacovigilance plan, the follow-up of efficacy and adverse reactions of any ATMP should systematically include the report of the ATMP batch number. In addition, access to registries should be detailed and patient informed consent in place. In comparison to “classical” medicinal products, additional risks of ATMPs are expected and concern quality characteristics, administration (sometimes extremely complicated), storage, and distribution of the GTMP. They also include not only those related to the severe underlying disease but also those linked to the persistence of the product within the patient’s body.
Therefore, the applicant should detail their strategy for identifying these risks, outline their approach for managing the reporting of adverse reactions (e.g., with or without follow-up questionnaires), and specify the proposed risk minimization measures, as well as post-authorization studies related to safety and/or efficacy.
For GTMPs, additional issues are extensively listed in the “Guideline on follow-up of patients administered with gene therapy”[8] with quality requirements, especially regarding viral vectors. Many reflection papers from EMA or other agencies are also proposed to discuss these quality, non-clinical and clinical issues and requirements more in-depth, some from EMA[9,10].
A high risk for delayed ADR particularly involves viral vectors mediating genetic material transfer into the cell nucleus and replicating (oncolytic) viruses. Additionally, vectors or viruses that remain in the cytoplasm or undergo abortive replication are associated with a low risk of malignant transformation. Unexplored risks necessitate a comprehensive evaluation that incorporates existing non-clinical and clinical data, along with the collective experience gained from other analogous GTMPs[8]. To exemplify this, we will delve into the case of betibeglogene autotemcel.
Vector-based GTMPs are potentially associated with the risk of insertional mutagenesis leading to oncogenesis[10-12]. The risk of virus-mediated transduction using recombinant retroviruses was evidenced in 1983 in mice[13]. Later, in clinical trials using therapeutic retroviral vectors, five of 20 patients who were treated for X-linked severe combined immunodeficiency disease developed leukemia[14].
The decision about the duration of the patient’s clinical follow-up is particularly challenging and is relevant to the assessment of the potential for chromosomal integration of a vector or a gene associated with the risk of malignant transformation of cells, which is related to insertional mutagenesis and activation or involves host cell genes inactivation or alteration. Integrating vectors such as gammaretroviral and lentiviral vectors, and vectors modified to induce integration allow permanent DNA integration, leading to long-term expression of corrective genes, and thus are especially concerned[12].
The duration of a patient’s clinical follow-up is also influenced by the capacity of the vector for latency or reactivation, or for inadvertent replication after complementation by viruses that cause escape from latency and reactivation. Replicating (oncolytic) viruses may always become latent after initial in vivo replication. MAH are therefore encouraged to find methods for detecting such occurrences of reactivation. Plasmids and non-viral vectors may possess limited integration capacity, yet they are suspected to sustain the long-term persistence of the gene and its expression, and therefore may be associated with a high risk for delayed adverse reactions, especially due to immunopathological mechanisms.
Regarding genetically modified cells, the likelihood of delayed ADR is linked not only to the vector used for the genetic modification of cells, but also to the nature of the gene product, the life span of the modified cells, and their biodistribution. The potential lifelong persistence of these cells poses a special risk for delayed effects contingent upon the integrated vector. Additionally, risks of oncogenesis, immunogenicity or vector reactivation should also be considered.
The risks associated with the persistence of the product in the patient should also be evaluated in terms of available rescue procedures or antidotes, taking into consideration their inherent risks, particularly concerning late complications such as malignancies and autoimmunity. If there is a possibility of re-administrating the GT product, it is crucial to explore the risks of immune reactions, anaphylaxis, and the development of neutralizing antibodies.
When assessing specific parent-child risks, it is essential to investigate potential factors such as the integration of transgenes into the germ line, genetic transformation of the germ line, and fetal transmission of vectors and biologically active substances, as well as cells and infectious agents. These recommendations currently reflect a precautionary principle, but to our knowledge, clinical data about this risk appears to be lacking. Another intriguing point to consider is the parents’ approval for treating their sick child. In cases of Down’s syndrome, some parents express mixed feelings. They seek to improve their child's quality of life while also seeking assurance regarding the safety and efficacy of any proposed GTMP before consenting to their administration[15].
Furthermore, breastfeeding should not be authorized without a thorough examination of breast milk contents.
All the available information enables the classification of the safety concerns into three categories concerning the GT product: important identified risks, important potential risks, and areas where information is lacking. In accordance with the current EU legislation and pharmacovigilance guidelines, these specifications will form an integral part of the RMP submitted alongside the application for marketing authorization. The RMP will evolve as new data from ongoing studies become available, enabling adjustments such as addressing missing information, redefining identified and potential risks, and ensuring the document stays updated.
Regarding delayed adverse reactions, which are particularly difficult to identify and diagnose, it is crucial to emphasize the importance of promptly reporting any suspected adverse reactions and seeking input from a pharmacovigilance expert.
Finally, the MAH of a GT medicinal product should retain, for a minimum of 30 years after the expiry date of the product (longer if required), all traceable data about its origin, manufacturing, packaging, storing, transportation, and delivery to healthcare facilities. Simultaneously, users must ensure the traceability of both the patient and the product.
GTPMS and safety signals
Overview
Currently, several GTMPs are authorized in the EU, USA, China, and other countries. Many of them already had their MA withdrawn for safety reasons, lack of efficacy, or by the MAH for commercial reasons [Table 1].
Non-exhaustive list of gene therapy medicinal products, safety signals and current marketing authorization status
| INN (specialty name) | Virus | Indication | Administration route | Safety signal | MA status |
| Viral vectors | |||||
| Alipogene tiparvovec (Glybera®) | Adenovirus | Familial lipoprotein lipase deficiency | Intramuscular | Lack of long-term safety data and insufficient efficacy | Expired in EU on 30/10/2017 |
| Beremagene geperpavec (Vyjuvek®) | HSV1 | Epidermolysis bullosa | Topical | Ongoing in the USA | |
| Contusugene ladenovec (Advexin®) | Adenovirus | Li-Fraumeni cancer | Injection | Lack of evidence on efficacy and safety | Withdrawn in EU on 10/09/2009 |
| Etranacogene dezaparvovec (Hemgenix®) | Adenovirus | Haemophilia B | Intravenous infusion | Ongoing MA in EU | |
| Lenadogene nolparvovec (Lumevoq®) | Adenovirus | Leber hereditary optic neuropathy | Intravitreal | Withdrawn in UE on 26/04/2023 Lack of evidence on efficacy | |
| Onasemnogene abeparvovec (Zolgensma®)* | Adenovirus | Spinal muscular atrophy | Intravenous infusion | Thrombotic microangiopathy Fatal acute liver failure | Ongoing in EU |
| Valactocogene roxaparvovec (Roctavian®) | Adenovirus | Haemophilia A | Intravenous infusion | Ongoing in EU | |
| Voretigene neparvovec (Luxturna®) | Adenovirus | Leber congenital amaurosis retinitis pigmentosa | Subretinal | Ongoing in EU | |
| Genetically modified cells | |||||
| Atidarsagene autotemcel (Libmeldy®) | Lentivirus | Metachromatic leukodystrophy | Intravenous infusion | Ongoing in EU | |
| Autologous CD34+ cells encoding ARSA gene (Strimvelis®) | Retrovirus | Severe combined immunodeficiency due to adenosine deaminase deficiency | Intravenous infusion | One case of lymphoid T cell leukemia after insertional oncogenesis | Ongoing in EU |
| Axicabtagene ciloleucel (Yescarta®) | Retrovirus | High-grade B-cell lymphoma Diffuse large B-cell lymphoma Primary mediastinal large B-cell lymphoma Follicular lymphoma | Intravenous infusion | Ongoing in EU | |
| Betibeglogene autotemcel (Zynteglo®)* | Lentivirus | β-thalassemia | Intravenous infusion | Suspicion of leukemia, not confirmed | Withdrawn in EU on MAH request on 7/08/2022 |
| Brexucabtagene autoleucel (Tecartus®) | Retrovirus | Mantle cell lymphoma Acute lymphoblastic leukemia | Intravenous infusion | Ongoing in EU | |
| Ciltacabtagene autoleucel (Carvykti®) | Lentivirus | Multiple myeloma | Intravenous infusion | Ongoing in EU | |
| Eivaldogène autotemcel (Skysona®) | Lentivirus | Adrenoleukodystrophy | Intravenous infusion | No patient treated in UE | Withdrawn in EU on MAH request for commercial reasons on 4/04/2022 |
| Exagamglogene autotemcel (Casgevy®) | No vector | Sickle-cell disease β-thalassemia | Intravenous infusion | Under evaluation in USA | |
| Idecabtagene vicleucel (Abecma®) | Lentivirus | Multiple myeloma | Intravenous infusion | Ongoing in EU | |
| Lisocabtagene maraleucel (Breyanzi®) | Lentivirus | Diffuse large B-cell lymphoma High-grade B-cell lymphoma Primary mediastinal large B-cell lymphoma Follicular lymphoma | Intravenous infusion | Ongoing in EU | |
| Nalotimagene carmaleucel (Zalmoxis®) | Retrovirus | Hematopoietic stem cell transplantation Graft vs. host disease | Intravenous infusion | Withdrawn in EU on MAH request for commercial reasons on 14/04/2020 | |
| Recombinant human p53 adenovirus (Gendicine®) | Adenovirus | Head and neck squamous cell carcinoma | Intralesional | Ongoing in China | |
| Relmacabtagene autoleucel (Carteyva®) | Lentivirus | Large B-cell lymphoma | Intravenous infusion | Ongoing in China | |
| Talimogene laherparepvec (Imlygic®) | HSV1 | Melanoma | Intralesional | Potential occurrence of adverse events in HSV-1 seronegative patients receiving vials with the less active product (less viral infectivity) | Ongoing in EU |
| Tisagenlecleucel (Kymriah®) | Lentivirus | B-cell acute lymphoblastic leukemia Diffuse large B-cell lymphoma | Intravenous infusion | Ongoing in EU | |
Among this permanently evolving landscape, we chose the two recent examples of safety signals raised for onasemnogene abeparvovec (Zolgensma®) and betibeglogene autotemcel (Zynteglo®) to illustrate the pharmacovigilance management with possible different outcomes.
Onasemnogene abeparvovec (Zolgensma®), thrombotic microangiopathy and acute liver failure
Onasemnogene abeparvovec (OA) is an adeno-associated virus (AAV)-based gene replacement therapy for treating spinal muscular atrophy (SMA). It aims to replace the defective primary SMN gene and is intended for patients diagnosed with SMA Type 1 due to a bi-allelic mutation in the SMN1 gene, or those with 5q SMA possessing a bi-allelic mutation in the SMN1 gene and up to three copies of the SMN2 gene. OA has been authorized for use throughout the European Union since May 18th, 2020. Two safety concerns associated with OA were thrombotic microangiopathy (TMA) and acute liver failure.
In preclinical studies performed in neonatal mice, piglets, and juvenile or neonatal cynomolgus nonhuman primates (NHPs), the main target organs of toxicity were the heart and liver in mice, and dorsal root ganglia (DRG). In mice, atrial thrombosis was often considered the cause of death, whereas no DRG inflammation was found. In cynomolgus NHPs, no thrombi were observed, but DRG mononuclear cell inflammation occurred, sometimes with neuronal satellitosis or necrosis after the intrathecal route of administration[16]. Regarding hepatotoxicity, severe toxicity occurred in both NHPs and piglets. All three NHPs had increased transaminases, two of whom recovered without sequelae, while one developed acute liver failure and shock, leading to euthanasia four days after a single OA intravenous administration[17].
During clinical studies, hepatotoxicity was confirmed. In 2020, Chand et al. evidenced liver-associated adverse events in 34% of 100 patients with SMA (without elevated baseline hepatic enzymes) who had received OA[18]. The liver function recovered after prednisolone, taking anywhere from 33 to 229 days, with most cases showing improvement within the range of 60 and 120 days. More than 40% of patients also had potentially hepatotoxic concomitant medications.
In 2021, Day et al. observed several cases in the post-marketing phase: four cases of TMA and 375 cases of hepatotoxicity[16]. Among the hepatotoxicity cases, 337 involved elevated liver function tests, while 14 cases were symptomatic with jaundice and/or ascites, and/or abnormal laboratory tests (coagulopathy, hypoalbuminemia, and increased ammonia), and four cases of acute liver failure. Among these four cases, two had been previously reported by Feldman et al.: the first involved a 6-month-old male who had received 3 doses of nusinersen and displayed increased AST and ALT (216 U/L and 234 U/L, respectively) before treatment[19]. He experienced acute liver failure approximately seven weeks after Zolgensma dosing. The second patient was a 20-month-old female who had received 8 doses of nusinersen and had normal baseline transaminases levels. She developed acute liver failure approximately 8 weeks after Zolgensma dosing. Liver biopsy showed acute inflammation, with portal abnormalities and mild interface hepatitis (mainly neutrophils, CD8+ T cells, and eosinophils), reactive bile ducts, but no acute cholangitis. Both patients recovered after high-dose steroid treatment.
In 2021, the regulatory safety assessment of OA totalized five confirmed cases of TMA in patients aged 4-23 months after treatment, among approximately 800 treated patients. Therefore, a DHPC was issued on March 18th, 2021 to alert pediatric neurologists, hematologists, and nephrologists about the risk of TMA, particularly during the initial weeks of the treatment. It emphasized the necessity to closely monitor creatinine levels and complete blood count before administration of OA, along with platelet counts in the week following infusion and regularly thereafter. Additionally, the DHPC stressed the importance of providing specific information to caregivers.
However, in August 2022, Novartis reported two fatal cases of acute liver failure[20]. The patients, aged 4 and 28 months, respectively, initially had an asymptomatic elevation of aminotransferases controlled with prednisolone dose. However, their clinical signs worsened, liver aminotransferases increased again, and the outcome was unfavorable, with rapid worsening of liver function, progression to hepatic encephalopathy and multi-organ failure. The two patients died around seven weeks after the OA infusion, during the period of corticosteroid dose tapering. These two fatal cases prompted the issuance of another DHPC on February 16th, 2023 to alert about the risk of fatal acute liver failure and recommend monitoring liver function both before treatment and regularly for at least 3 months after infusion. Considering the progression of the two cases, it was also recommended not to start corticosteroid tapering until liver function tests were normalized alongside a normal clinical examination.
The mechanism behind hepatotoxicity seems related to an innate and/or adaptive immune response to the AAV vector. There is a strong suspicion that the transduction of the AAV vector and its subsequent expression in Kupffer cells may activate capsid-specific T cells once a certain threshold of capsid antigen load is reached. This activation could result in hepatotoxicity[18]. Additionally, it is worth noting that patients with SMA and mouse models of SMA show an increased risk of dyslipidemia, fatty acid metabolism defects, and steatosis, thus probably contributing to hepatotoxicity of OA[19].
Betibeglogene autotemcel (Zynteglo®) and suspicion of insertional oncogenesis leading to leukemia
Betibeglogene autotemcel (BA) is a potential curative treatment of β-thalassemia (BT), a heterogeneous autosomal recessive hereditary anemia due to reduced or absent β-globin chain synthesis. Patients with BT major have severe anemia shortly after birth. In the absence of β-Globin, α-Globin tetramers accumulate and precipitate in the red blood cells (RBC) precursors, forming inclusion bodies leading to oxidative membrane damage and ineffective erythropoiesis. BT management is based on chronic transfusions, generating varied complications, such as secondary hemochromatosis. Other solutions are bone marrow transplant, iron chelation therapy, and splenectomy.
The BA treatment involves a genetically modified autologous CD34+ cell-enriched population that contains hematopoietic stem cells (HSC) transduced with a lentiviral vector (LVV) encoding the β A-T87Q-globin gene. Authorized in May 2019, this treatment is intended for patients aged 12 years and older with transfusion-dependent β-thalassemia (TDT). It specifically targets patients whose genotype differs from β0/β0 and who have no available HLA-matched related hematopoietic stem cell transplantation donor. The patient undergoes one or more autologous hematopoietic stem cell mobilization cycles to obtain CD34+ stem cells. The harvested cells are transduced ex vivo with self-inactivating lentiviral vectors, which insert the globin gene and other elements required for expression. In addition to common Grade 3 or 4 laboratory abnormalities (> 50%) (neutropenia, thrombocytopenia, leukopenia, anemia, and lymphopenia), BA safety concerns include insertional oncogenesis leading to hematologic malignancies, and hypersensitivity reactions due to dimethyl sulfoxide (DMSO). As full myeloablative conditioning is required before infusion, its inherent risks (cytopenia, infertility, and secondary malignancy) should also be taken into consideration before choosing this treatment.
From a regulatory standpoint, the CHMP recommended in September 2021 the renewal of the conditional marketing authorization of BA (Zynteglo®). However, on February 18th 2021, the European Commission (EC) triggered a procedure for the review of BA after being aware of a case of acute myeloid leukemia in a patient with sickle-cell disease receiving bb1111, a GT investigational product using the same lentiviral vector as BA. In addition, two other patients who received bb1111 had developed myelodysplastic syndrome, which progressed to leukemia in one of them. Thus, the lentiviral vector was suspected to exert insertional oncogenesis[21]. The CHMP was requested to assess the impact of this information on the benefit-risk balance of Zynteglo. As this new data resulted from pharmacovigilance activities, the CHMP based its opinion on a recommendation of the PRAC, supported by experts from the CAT, who concluded that the viral vector was unlikely to be the cause. This was based on the findings that i) one patient did not have the viral vector in his cancer cells, and ii) for the other patient, the viral vector was present at a site (VAMP4) not involved in cancer development. Finally, the conditioning myeloablative treatment and the expected risk of blood cancer in people with sickle cell disease were considered more plausible explanations for the AML cases. Nevertheless, despite this favorable conclusion, the EC withdrew the marketing authorization for Zynteglo in the EU on 24 March 2022 at the request of the MAH, which decided to permanently discontinue it for commercial reasons[22]. These reasons included manufacturing issues, which had forced the company to delay Zynteglo’s launch for many months, and the failure of price negotiations with health authorities.
CONCLUSION
PV activities and systems must constantly adapt to evolving regulations and the advancement of new therapies and technologies. At present, gene therapy, with its promising therapeutic issues, represents probably one of the most challenging pharmacovigilance domains. Indeed, besides already identified and potential safety concerns, GTMPs can also generate unprecedented security issues and potential complexities that will require high skills and probably substantial time to resolve. Therefore, at each level, GTMPs PV necessitates the active participation of proficient pharmacological and clinical experts across various domains, such as pharmaceutical sciences, biostatistics, epidemiology, and sociology, to effectively address emerging challenges and achieve the best outcomes. GTMPs PV requires the comprehensive engagement of healthcare professionals, MAHs, and patients to establish robust pharmacovigilance plans, facilitate the reporting of adverse reactions, ensure transparent assessments, and optimize the management of genetic diseases and other areas that can benefit from gene therapy. As for any medication, the success of GTMPs PV relies on collective commitment and concerted efforts from all stakeholders.
DECLARATIONS
Author’s contributions
Collected information and drafted the article: Petitpain N, Fresse A, Antoine ML
Reviewed the article: Beurrier M, Micallef J
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflict of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
1. Orzetti S, Tommasi F, Bertola A, et al. Genetic therapy and molecular targeted therapy in oncology: safety, pharmacovigilance, and perspectives for research and clinical practice. Int J Mol Sci 2022;23:3012.
2. World Health Organization. The importance of pharmacovigilance. Available from: https://www.who.int/publications-detail-redirect/10665-42493 [Last accessed on 20 Nov 2023].
3. Kugener VF, Freedland ES, Maynard KI, et al. Enhancing pharmacovigilance from the US experience: current practices and future opportunities. Drug Saf 2021;44:843-52.
4. EMA European Medicine Agency. Good pharmacovigilance practices. Available from: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/pharmacovigilance/good-pharmacovigilance-practices#final-gvp-annex-i---definitions-section [Last accessed on 20 Nov 2023].
5. EMA European Medicine Agency. Guideline on good pharmacovigilance practices (GVP) module XV - safety communication (Rev 1). Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-xv-safety-communication-rev-1_en.pdf [Last accessed on 20 Nov 2023].
6. EMA. How the committees work. Available from: https://www.ema.europa.eu/en/committees/how-committees-work [Last accessed on 20 Nov 2023].
7. EMA European Medicine Agency. Guideline on safety and efficacy follow-up and risk management of advanced therapy medicinal products-scientific guideline. Available from: https://www.ema.europa.eu/en/guideline-safety-efficacy-follow-risk-management-advanced-therapy-medicinal-products-scientific [Last accessed on 20 Nov 2023].
8. EMA European Medicine Agency. Guideline on follow-up of patients administered with gene therapy medicinal products. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-follow-patients-administered-gene-therapy-medicinal-products_en.pdf [Last accessed on 20 Nov 2023].
9. EMA European Medicine Agency. Reflection paper on quality, non-clinical and clinical issues related to the development of recombinant adenoassociated viral vectors. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-quality-non-clinical-clinical-issues-related-development-recombinant-adeno_en.pdf [Last accessed on 20 Nov 2023].
10. EMA European Medicine Agency. Reflection paper on management of clinical risks deriving from insertional mutagenesis. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-management-clinical-risks-deriving-insertional-mutagenesis_en.pdf [Last accessed on 20 Nov 2023].
11. David RM, Doherty AT. Viral vectors: the road to reducing genotoxicity. Toxicol Sci 2017;155:315-25.
12. Aiuti A, Cossu G, de Felipe P, et al. The committee for advanced therapies' of the European Medicines Agency reflection paper on management of clinical risks deriving from insertional mutagenesis. Hum Gene Ther Clin Dev 2013;24:47-54.
13. Wagner EF, Covarrubias L, Stewart TA, Mintz B. Prenatal lethalities in mice homozygous for human growth hormone gene sequences integrated in the germ line. Cell 1983;35:647-55.
14. Hacein-Bey-Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 2010;363:355-64.
15. Riggan KA, Nyquist C, Michie M, Allyse MA. Evaluating the risks and benefits of genetic and pharmacologic interventions for down syndrome: views of parents. Am J Intellect Dev Disabil 2020;125:1-13.
16. Day JW, Mendell JR, Mercuri E, et al. Clinical trial and postmarketing safety of onasemnogene abeparvovec therapy. Drug Saf 2021;44:1109-19.
17. Hinderer C, Katz N, Buza EL, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther 2018;29:285-98.
18. Chand D, Mohr F, McMillan H, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol 2021;74:560-6.
19. Feldman AG, Parsons JA, Dutmer CM, et al. Subacute liver failure following gene replacement therapy for spinal muscular atrophy type 1. J Pediatr 2020;225:252-8.e1.
20. NOVARTIS. Zolgensma acute liver failure update. Available from: https://www.novartis.com/news/zolgensma-acute-liver-failure-update [Last accessed on 20 Nov 2023].
21. EMA European Medicine Agency. Pharmacovigilance risk assessment committee (PRAC) PRAC minutes on 08-11 March 2021. Available from: https://www.ema.europa.eu/en/documents/minutes/minutes-prac-meeting-8-11-march-2021_en.pdf [Last accessed on 20 Nov 2023].
22. EMA European Medicine Agency. Zynteglo withdrawal of the marketing authorisation in the European Union. Available from: https://www.ema.europa.eu/en/documents/public-statement/public-statement-zynteglo-withdrawal-marketing-authorisation-european-union_en.pdf [Last accessed on 20 Nov 2023].
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Petitpain N, Antoine ML, Beurrier M, Micallef J, Fresse A. Pharmacovigilance of gene therapy medicinal products. Rare Dis Orphan Drugs J 2023;2:25. http://dx.doi.org/10.20517/rdodj.2023.19
AMA Style
Petitpain N, Antoine ML, Beurrier M, Micallef J, Fresse A. Pharmacovigilance of gene therapy medicinal products. Rare Disease and Orphan Drugs Journal. 2023; 2(4): 25. http://dx.doi.org/10.20517/rdodj.2023.19
Chicago/Turabian Style
Petitpain, Nadine, Marie Lauren Antoine, Mathilde Beurrier, Joelle Micallef, Audrey Fresse. 2023. "Pharmacovigilance of gene therapy medicinal products" Rare Disease and Orphan Drugs Journal. 2, no.4: 25. http://dx.doi.org/10.20517/rdodj.2023.19
ACS Style
Petitpain, N.; Antoine ML.; Beurrier M.; Micallef J.; Fresse A. Pharmacovigilance of gene therapy medicinal products. Rare. Dis. Orphan. Drugs. J. 2023, 2, 25. http://dx.doi.org/10.20517/rdodj.2023.19
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 4
likes
Like This Article 4
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.