
The Australian landscape of newborn screening in the genomics era


Abstract
In Australia, over 300,000 newborns undergo newborn bloodspot screening (NBS) annually, with approximately 1 in 1,000 identified with a rare but actionable condition through this pathway. Prior to 2018, the inclusion criteria for adding conditions in NBS panels was inconsistent nationally, leading to the development of the Australian National Newborn Bloodspot Screening Policy Framework. This framework promotes systematic and evidence-based inclusion of conditions using criteria closely informed by traditional Wilson and Junger screening principles. Current policy initiatives are focused on achieving national consistency in the conditions screened. NBS programs, initiated in the 1960s, have used a variety of techniques, including but not limited to tandem mass spectrometry and immunological assays. The acceleration of genomic technologies has the potential to greatly increase the number of conditions screened and match affected newborns with innovative treatment options, including advanced (gene, immune modulation, and RNA) therapies. This review describes the evolution, current status quo, and outlook for Australian NBS programs with a focus on the implications of wider adoption of genomic newborn screening (gNBS) in our culturally, geographically, and genetically diverse population. We discuss the potential for transformative benefits for families with children identified by gNBS and how this must be balanced against the potential for a range of unintended negative consequences. We emphasise the importance of a nationally agreed, coordinated, and streamlined approach to the addition and removal of conditions from Australian NBS programs, which considers the utility, cost, ethical, and equity aspects of gNBS.
Keywords
INTRODUCTION
NBS is a public health program that aims to detect newborns with serious, actionable disorders to enable timely and appropriate interventions and improve health outcomes. In Australia, NBS is a well-established program, screening for over 50 conditions, and has been offered to all newborns since the 1960s[1,2]. Australian NBS is associated with high levels of public trust, with a > 99% uptake rate nationally in 2023, 300,000 newborns screened every year, and approximately 1 in 1,000 babies identified with a rare condition that would otherwise have gone undetected until symptoms arose[2].
The inclusion of specified conditions into routine Australian NBS programs was traditionally informed by the Population Based Screening Framework[3], which remains closely aligned with the internationally recognised ten screening principles developed by Wilson and Jungner and endorsed by the World Health Organisation (WHO) in 1968[4] (Box 1). Prior to 2018, the addition of new conditions into state and territory NBS panels was assessed within each jurisdiction's infrastructure. Evaluation of net benefits for NBS of rare disorders necessitated large pilot studies to accrue sufficient evidence of alignment with screening principles before endorsement of a condition onto routine NBS panels. This approach led to variable timing of implementation between health jurisdictions within Australia, resulting in inequity in access to diagnosis and specialist care[5,6]. Changes in policy to enable best practice through the development of a Newborn Screening Policy Framework in 2018 were driven in part by the imperative to mitigate these inequities[7].
Box 1. Original Wilson and Jungner principles for population-based screening
Derived from “Principles and Practice of Screening for Disease”[4]
The condition should be an important health problem
There should be an accepted treatment for patients with the disease
Facilities for diagnosis and treatment should be available
There should be a recognizable latent or early symptomatic stage
There should be a suitable test or examination
The test should be acceptable to the population
The natural history of the condition, including development from latent to declared disease, should be adequately understood
There should be an agreed policy on whom to treat as patients
The cost of case-finding (including diagnosis and treatment of patients diagnosed) should be economically balanced in relation to possible expenditure on medical care as a whole
Case-finding should be a continuing process and not a "once and for all" project
On the technology front, rapid advances in genomic sequencing capabilities and gene- and RNA- based treatments are driving the emergence of new opportunities and challenges for Australian NBS programs. The ability to perform DNA extraction on dried blood spots (DBS) at scale has facilitated the incorporation of first-tier genetic screening tests for conditions such as spinal muscular atrophy (SMA), with Australia one of only nine countries globally to recommend the inclusion of SMA in NBS programs[8]. The opportunity to change the disease course of these predominantly serious, childhood-onset conditions through earlier diagnosis and treatment, ameliorating the substantial health burden on affected individuals, families, and health systems, has been cited as a potential benefit of an expanded NBS program[9-12].
Beyond single gene tests, a range of new modalities for NBS are emerging within the research domain and being evaluated as either complementary or adjunctive to existing NBS pathways.
These include the capacity to incorporate next-generation genomic sequencing techniques, including whole exome (WES) and genome sequencing (WGS)[13], methylation studies for imprinting disorders[14], and metabolomic biomarkers for profiling of people who are at increased risk of serious and actionable disorders[15].
These emerging screening opportunities also underpin interest among stakeholders to expand NBS to include conditions or genetic variants that do not necessarily fulfil the traditional principles of newborn population screening, such as conditions with an older age of onset or the use of genomic profiles to predict future risks of a condition[16-18]. However, if we are to utilise gNBS in a way that is equitable, effective, cost-effective, and ethically informed, we need to ask not only “can we use genomics to screen newborns”? but also: “should we use genomics to screen newborns” and “what are we as a society prepared to pay for that screening”? These questions are important when considered in the context of an existing effective screening program with a high level of uptake and public trust.
Within this context, this paper provides an overview of the evolution, current state, and outlook for NBS programs in Australia. Considering the incorporation of genomic technologies within existing NBS, we also explore ways in which the current Australian NBS National Policy Framework may need to evolve and adapt if it is to account for the full range of primary and secondary benefits associated with the early treatment of a much higher number of rare and ultra-rare conditions[19-22].
NEWBORN BLOODSPOT SCREENING ORGANISATION AND COORDINATION IN AUSTRALIA
The organisation of NBS programs in Australia reflects the country’s federated system of government, with eight jurisdictional governments (representing 6 States and 10 Territories) and a national Commonwealth government. As health is a devolved power in Australia’s constitution, the implementation of NBS programs is the responsibility of the state and territory governments.
There are 5 Australian NBS reference centres (located in Adelaide, Brisbane, Melbourne, Perth, and Sydney) providing coordination of NBS programs[23]. These laboratories screen DBSs collected onto filter paper, taken from the newborn’s heel ideally 48-72 h from birth. Each DBS contains three unique patient identifiers and a named paediatrician for contact. The consent process for the collection of DBSs typically includes a verbal description of the test and its benefits, a pamphlet, and, in some jurisdictions, a guide to a web-based resource. The Australian NBS program is not mandatory, and parents can opt out of the screening test[2].
Funding for healthcare in Australia is derived from a mix of public and private sources, with the majority (70.6%) of healthcare funded by the government[24]. While all formal population screening programs in Australia, including NBS programs, are publicly funded with no out-of-pocket costs for the screened individual, pathways to care for screen-positive individuals are highly variable depending on their knowledge of and access to public and/or private health services. Variable access to health services is a common challenge in Australia and arises as a consequence of the relatively small population (25.7 million) spread across a large geographical area (7.7 million km2) with wide diversity in health literacy, socio-economic circumstances, language, and cultural perspectives[25]. On rare occasions, these challenges give rise to missed screening opportunities, such as for some babies born at home, in remote/regional areas, families with sporadic antenatal care, or mothers who normally reside in other states[26]. A very small proportion of parents decline screening for their babies. More frequently, though, challenges with accessing appropriate care are apparent in referral pathways for newborns and children diagnosed with rare conditions, as specialist services required for care tend to be in a limited number of major metropolitan centres[27,28]. Access to follow-up care is one of the key equity challenges for Australia as we consider how to expand existing NBS programs.
The drivers of newborn bloodspot screening policy change in Australia
Prior to 2018, individual states and territories in Australia determined their own screening targets, leading to differences between jurisdictions in the conditions screened. From the 1980s, the five screening laboratories in Australia voluntarily governed themselves through a joint committee of The Royal Australasian College of Physicians and the Human Genetics Society of Australasia (HGSA). As new technologies became available and inequities between programs arose both in the form of conditions screened and access to specialist care and support, it became increasingly clear that broader community engagement and a national approach were required. In response, the HGSA sought guidance from the federal government and an expert working group was formed to develop a cohesive NBS framework as a step toward a standardised approach to NBS nationally, with the federal government launching the Newborn Bloodspot Screening: National Policy Framework in 2018[7]. The Framework provided a series of considerations to defer to when jurisdictions were determining whether to add, or remove, a condition for screening. For the first time, it allowed a mechanism for harmonising processes for managing identified children, program monitoring and evaluation, consent, storage oversight, and evidence-based program expansion (or reduction, as appropriate)[7,29].
However, it should be noted that at the time the Framework was developed, no additional funds were allocated by the government to support the assessment of new conditions or implementation of expanded screening. Since then, in response to patient advocacy around remaining inequities in access to screening, the Australian government formally launched the Newborn Screening Infrastructure in late 2022, with a monetary commitment of $AU 39.4 million over four financial years. The purpose of this investment is to enable the delivery of an optimised, equitable, and integrated screening program, including the implementation of regular processes to update, synchronise, and expand (or remove) screened conditions in line with international best practices[30]. The nationally coordinated approach will also support increased funding for treatment pathways for actionable rare conditions identified through NBS[31]. Although this support has been welcomed by the sector, further refinements to the assessment pathway for NBS conditions are still required, especially against the backdrop of wider use of gNBS, and questions remain regarding ongoing funding beyond the current commitment.
THE EVOLUTION OF NEWBORN BLOODSPOT SCREENING TECHNOLOGIES IN AUSTRALIA
NBS in the tandem mass spectrometry (tMS) age included screening for conditions of amino acid, organic acid, and fatty acid oxidation metabolism, with Australia being a leader among international tMS programs[32]. The era of tMS was perhaps the first time when well-established screening principles had been challenged, with the possibility of adding a myriad of conditions (pathogenic and non-pathogenic) onto the NBS panel with a negligible increase in the cost of case finding.
The pitfalls of “counting of conditions” in newborn bloodspot screening programs
The naming and counting of conditions on NBS panels varies between programs nationally and internationally. This is related to a range of factors, including the nomenclature used to identify conditions and the emergence of systems that count screened tMS analytes versus identified conditions. This has led to varying perceptions of how many conditions were being screened within each program and questionable comparisons between programs as to “best practice”[32-38] [Figure 1].

Figure 1. (A) Newborn bloodspot screening for methylmalonic acidaemia and propionic acidaemia demonstrates the differences in counting conditions by pathogenic variants or target analytes[36]. Pathogenic variations in nine different genes result in the elevation of a single analyte such as propionylcarnitine. Although this analyte was primarily used to detect methylmalonic acidaemia caused by pathogenic variation of the MMUT gene or propionic academia related to genetic variants in PCCA and PCCB genes, methylmalonic acidaemia caused by pathogenic variants in MMAA, MMAB, MMADHC, MMACHC, LMBRD1, and ABCD4 could also be identified; (B) Newborn bloodspot screening for the target analyte 3-hydroxyisovaleryl-/2-methyl-3-hydroxy acylcarnitine (C5-OH) may identify both Category 1 “target disorders” and Category 2 “incidental findings” when elevated. Category 1 and 2 disorders categorised and defined by The Human Genetics Society of Australasia (HGSA)[1]. Elevated C5-OH can lead to the identification of four “target disorders” and three “incidental findings.” Uncertainty exists surrounding the actionability of the latter “incidental findings”[37,38]. *Biotinidase deficiency is screened for in New Zealand but not in Australia. Cbl: cobalamin. This figure was created using https://www.biorender.com/.
Australian programs continue to focus on conditions that are medically actionable. For instance, while ornithine transcarbamylase (OTC) deficiency, an X-linked urea cycle disorder, is included in international routine NBS panels, the fact that males with the condition often present and require urgent treatment within the first days of life (before DBS samples can be collected and analysed), precluding this as an actionable condition to be screened for with Australian NBS programs[39]. To set Australian NBS programs within the scope of international best practice, the HGSA has provided recommendations on how disorders are classified [Table 1][1]. These recommendations are consistent with requests by the Australian rare disease community, which advocates for meaningful and consistent patient- and family-centred terms and language, rather than a focus on “counting of conditions” to guide best practice.
Comparison of conditions included in Australian and Californian newborn bloodspot screening programs[1,37,40-43]
| Condition | Inclusion in Australian NBS | Inclusion in Californian NBS, incorporation in the RUSP | Comments relevant to NBS analyses and treatment |
| (A) Category 1 “target” conditions as classified by the Human Genetics Society of Australia (HGSA) | |||
| Inborn errors of metabolism | |||
| 3-Hydroxy-3-Methylglutaric aciduria | Yes | Yes, RUSP core condition | |
| Argininosuccinic aciduria | Yes | Yes, RUSP core condition | |
| Carnitine uptake defect | Yes | Yes, RUSP core condition | |
| Carnitine acylcarnitine translocase deficiency | Yes | Yes, RUSP secondary condition | Classical forms typically present before NBS results reported |
| Carnitine Palmitoyltransferase I deficiency | Yes | Yes, RUSP secondary condition | Very rare in Australia |
| Carnitine Palmitoyltransferase II deficiency | Yes | Yes, RUSP secondary condition | Typically but not always late onset |
| Citrullinemia type I | Yes | Yes, RUSP core condition | |
| GAII (multiple acyl-CoA-dehydrogenase deficiency) | Yes | Yes, RUSP secondary condition | Diagnosed by acylcarnitine profile |
| (Classic) galactosemia | Yes | Yes, RUSP core condition | Not currently screened in the state of Victoria-being implemented |
| Other galactosemias (epimerase, kinase, mutarotase deficiencies) | Yes | No | |
| Glutaric acidemia type I | Yes | Yes, RUSP core condition | |
| Holocarboxylase synthase deficiency | Yes | Yes, RUSP core condition | |
| Homocystinuria (cystathionine beta-synthase deficiency) | Yes | Yes, RUSP core condition | Variable methods-methionine most common primary marker |
| Isovaleric acidemia | Yes | Yes, RUSP core condition | |
| β-Ketothiolase deficiency | Yes | Yes, RUSP core condition | |
| Long-chain L-3-Hydroxyacyl-CoA dehydrogenase deficiency | Yes | Yes, RUSP core condition | |
| Maple syrup urine disease | Yes | Yes, RUSP core condition | |
| Medium-chain Acyl-CoA dehydrogenase deficiency | Yes | Yes, RUSP core condition | |
| Methylmalonic acidemia (MMA-CoA Mutase) | Yes | Yes, RUSP core condition | |
| Methylmalonic acidemia (Cobalamin A&B disorders) | Yes | Yes, RUSP core condition | C3 elevation identifies several disorders |
| Methylmalonic acidemia (Cobalamin defects C, D v2) | Yes | Yes, RUSP secondary condition | C3 elevation identifies several disorders |
| Remethylation defects (MTHFR, MTR, MTRR, Cbl D v1) | Yes | Yes, although not included on the RUSP | Not consistent in Australia. Technical advice sought |
| Phenylketonuria - (PAH and pterin enzyme deficiencies) | Yes | Yes, RUSP core condition | Pterin disorders can be identified by newborn screening-further tests |
| Propionic acidemia | Yes | Yes, RUSP core condition | |
| Trifunctional protein Deficiency | Yes | Yes, RUSP core condition | |
| Tyrosinemia type II and III | Yes | Yes, RUSP secondary condition | |
| Very long-chain Acyl-CoA dehydrogenase deficiency | Yes | Yes, RUSP core condition | |
| Endocrine, immune and neuromuscular disorders | |||
| Congenital adrenal hyperplasia (21-hydroxylase deficiency) | Yes | Yes, RUSP core condition | |
| Cystic fibrosis | Yes | Yes, RUSP core condition | |
| Primary congenital hypothyroidism | Yes | Yes, RUSP core condition | |
| Severe combined immunodeficiencies | Yes | Yes, RUSP core condition | Being implemented in Australia |
| Spinal muscular atrophy | Yes | Yes, RUSP core condition | Being implemented in Australia |
| (B) Category 2 “incidental” findings as classified by the Human Genetics Society of Australia (HGSA) | |||
| 2-Methyl-3-Hydroxybutyric aciduria | No | Yes, RUSP secondary condition | Data unclear on childhood treatment |
| 2-Methylbutyrylglycinuria | No | Yes, RUSP secondary condition | Data unclear on childhood treatment |
| 3-Methylcrotonyl-CoA carboxylase deficiency | Yes | Yes, RUSP core condition | Several asymptomatic mothers ascertained from NBS |
| 3-Methylglutaconic aciduria (3MGA) | Yes | Yes, RUSP secondary condition | Type I can be identified by C5OH, but treatment is unclear; other forms are non-specific |
| Benign hyperphenylalaninemia | Yes | Yes, RUSP secondary condition | Not treated in childhood-managed for maternal PKU |
| Citrullinemia type II | Yes | Yes, RUSP secondary condition | Data unclear on childhood treatment |
| Congenital adrenal hyperplasia (11βMonooxygenase Deficiency) | Yes | Yes, although not included on the RUSP | 17-hydroxyprogesterone primary marker for 21-hydroxylase deficiency |
| Duarte galactosemia | No | Yes, RUSP secondary condition | Considered a benign variation |
| Ethylmalonic encephalopathy | No | Yes, although not included on the RUSP | Data unclear on childhood treatment. Isolated C4 elevation not investigated |
| Formiminoglutamic acidemia | No | Yes, although not included on the RUSP | Data unclear on childhood treatment. |
| Hypermethioninemia | No | Yes, RUSP secondary condition | Sparse long-term data with treatment |
| Isobutyrylglycinuria | No | Yes, RUSP secondary condition | Isolated C4 elevation not investigated |
| Malonic acidemia | Yes | Yes, RUSP secondary condition | Some cases have been identified by NBS and treated in Australia |
| Medium/short-chain L-3-HydroxyacylCoA dehydrogenase deficiency | No | Yes, RUSP secondary condition | Not currently screened in Australia |
| Short chain Acyl-CoA dehydrogenase deficiency (SCADD) | No | Yes, although not included on the RUSP | Over ascertained in screen population. Isolated C4 elevation not systematically investigated |
| T-cell related lymphocyte deficiencies | Yes | Yes, RUSP secondary condition | Incidental but actionable |
| Tyrosinemia type I | Yes | Yes, RUSP core condition | Considered primary NBS, but tyrosine is not reliable for diagnosing type I. Succinyl acetone is the best marker |
| Tyrosinemia, transient | No | No | Incidental finding-not treated. |
| Vitamin B12 deficiency | Yes | No | Incidental-elevation of C3 |
| X-linked agammaglobulinaemia | Yes | Yes, RUSP secondary condition | Incidental but actionable |
| (C) Conditions included on the Californian newborn bloodspot screening panel which are not screened for in Australia | |||
| Argininemia | Yes, secondary condition screened | Arginine not screened | |
| Biotinidase** | Yes, RUSP core condition | ||
| Carbamoylphosphate synthetase deficiency (CPS) | Yes, although not included on the RUSP | Low citrulline difficult to detect-classical cases present before day 7 | |
| GAMT (guanidinoacetate methyltransferase) deficiency | |||
| Glycogen storage disease Type II (Pompe)* | Yes, RUSP core condition | ||
| Gyrate atrophy of the choroid and retina | Yes, although not included on the RUSP | Ornithine not measured-slowly progressive disorder-later onset. | |
| Haemoglobinopathies* | Yes, RUSP core condition | ||
| Hyperornithinemia-Hyperammonemia-Homocitrullinuria syndrome | Yes, although not included on the RUSP | Rare disorder. Ornithine not measured | |
| Hyperprolinemia type I | Yes, although not included on the RUSP | Treatment unclear-Proline not measured | |
| Hyperprolinemia type II | Yes, although not included on the RUSP | Treatment unclear-Proline not measured | |
| Mucopolysaccharidosis type I* | Yes, RUSP core condition | ||
| Mucopolysaccharidosis type II* | Yes, RUSP core condition | ||
| Ornithine Transcarbamylase Deficiency (OTC) | Yes, although not included on the RUSP | Low citrulline difficult to detect–classical cases present before day 7 | |
| X Linked adrenoleukodystrophy# | Yes, RUSP core condition | ||
Incorporating genetics within existing newborn bloodspot screening programs
Beyond tMS, first-tier genetic testing for selected monogenic conditions has been recommended for national incorporation into Australian NBS programs since July 2022, after the successful state-wide implementation of pilot programs for SMA and primary immunodeficiencies in New South Wales (NSW) and the Australian Capital Territory (ACT).
The effectiveness of NBS, however, goes beyond the availability of an accurate test. Regardless of the ability of genetic testing to identify at-risk newborns, the success of screening for a particular condition remains intricately linked with processes for diagnosis and healthcare delivery. This is evidenced by the Australian experience of integration of NBS for SMA into current healthcare systems [Figure 2]. Here, the co-development of an efficient referral pathway and model of care has been imperative to facilitate equitable SMA diagnosis and optimise opportunities for early intervention, specialist, and social care to mitigate health inequities due to financial, geographical, cultural, or linguistic barriers[28].
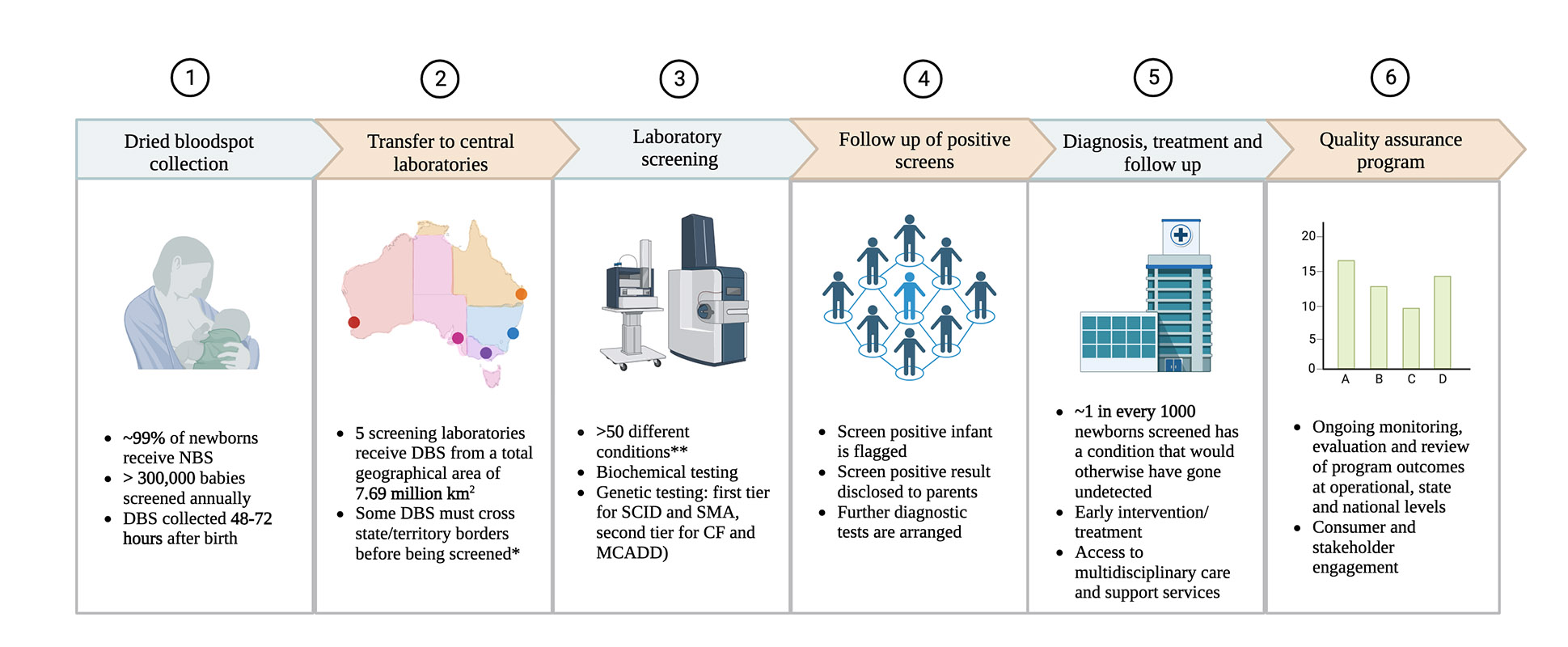
Figure 2. Integrated newborn bloodspot screening processes in Australia[7,44]. *In addition to dried blood spots (DBSs) collected from its own state, the Queensland (QLD) laboratory also receives cards from Katherine, Northern Territory (NT), and towns in NT north of Katherine. Similarly, the South Australian (SA) laboratory receives DBS cards from SA as well as Tasmania and towns south of Katherine in NT. **As per the Human Genetics Society of Australasia (HGSA)’s definitions and categorisation of conditions[1]. NBS: newborn bloodspot screening; DBS: dried bloodspot; SCID: severe combined immunodeficiency; SMA: spinal muscular atrophy: CF: cystic fibrosis: MCADD: medium-chain acyl-CoA dehydrogenase deficiency. This figure was created using https://www.biorender.com/.
Lessons learnt from the recent NBS for SMA program include challenges with delays in establishing an evidence base for NBS in rare conditions and developing the infrastructure on a state-by-state basis to incorporate genetic first-tier screening within existing NBS services. These challenges have led to current disparities in diagnostic and therapeutic interventions between newborns across Australia[45,46]. Babies born in NSW/ACT since July 2022 have been screened for SMA through NBS, while other states and territories are at various stages of implementing this screening. This experience has underlined the importance of horizon scanning for emergent therapies for rare conditions (a key factor in the Ministerial decision to recommend screening for SMA), so that the future expansion of Australian NBS programs is better coordinated with the availability of new therapies that are safe, effective and cost-effective[25,47].
FUTURE OUTLOOKS FOR NEWBORN BLOODSPOT SCREENING IN AUSTRALIA
Positioning Australia to evaluate the role of genomics in newborn bloodspot screening for rare conditions
Rare conditions are estimated to affect around 2 million Australians. Of these, many are genetic and the majority (82%) have childhood onset[48]. The National Strategic Action Plan for Rare Diseases, launched in February 2020 with bipartisan Commonwealth government support, was the first nationally coordinated effort to address the substantial cumulative health impact of these rare conditions. Among the Action Plan’s key priorities were the development and integration of genomics into healthcare to enable Australians to have equitable access to the best available health technology[49]. Since this time, NBS programs have become a key focus area for such integration.
In 2021, there was a targeted call for applications within a specified stream of the Genomics Health Futures Mission of the Medical Research Futures Fund looking ahead to the possible incorporation of gNBS. This called for projects to develop new models of NBS that incorporated genomics, either as a first-line test or a complement to existing biochemical screening[50]. Five projects, which engage through the Genomic Screening Consortium for Australian Newborns (GenSCAN)[51], have been funded under this call and are now underway. These are exploring the feasibility, scalability, and cost-effectiveness of gNBS, as well as its ethical, legal, and social aspects- including acceptability for Australia’s Indigenous populations and those from culturally, ethnically, and linguistically diverse groups. Scientific aspects being studied include the validity and utility of newborn genome sequencing, the use of epigenomics to identify newborns at risk of imprinting disorders, and metabolomic profiling on DBS to stratify newborns at risk of metabolic conditions into further screening opportunities. The projects are also considering which conditions to screen for, including how this can reflect condition prevalence in diverse populations and draw on appropriate genomic datasets for variant calling and interpretation.
The impetus for these projects has come in part from the significant increase in the use of genomic sequencing, including WES and WGS, in clinical practice and translational research. This, in turn, has been driven by a decline in sequencing costs and improved time to results[52,53]. Such testing has become the diagnostic standard of care for children with a suspected monogenic condition, demonstrating diagnostic yields of up to 50% in some Mendelian cohorts[54]. In Australia, WES is approved and reimbursed for diagnostic investigation of children with moderate to severe intellectual disability, and there is evidence of effectiveness in the acute care setting[55]. While WES identifies variants in the protein-coding areas and intronic regulatory sequences of the nuclear genome, WGS has the added benefit of detecting structural rearrangements, copy number variants, non-coding regions, and mitochondrial DNA. Currently used short-read technologies cannot detect disorders of methylation (which cause imprinting syndromes), but long-read technologies will be able to do so. Importantly, WES and WGS can miss important genetic information, such as pathogenic tandem repeat expansions[56]. gNBS may also have the challenge of revealing variants of unknown significance (VUS) or unsolicited findings (UFs), although these can be avoided through a well-defined gene list and careful variant curation, as has occurred in other settings where genomics has been modelled in a screening setting, such as the Mackenzie’s Mission project for reproductive genetic carrier screening (RGCS)[57].
BUILDING READINESS IN AUSTRALIA FOR THE NEXT GENERATION OF NEWBORN BLOODSPOT SCREENING MODALITIES
There is a strong consensus that while gNBS is not currently ready for integration into the Australian healthcare system, it should and could be implemented within the next decade[25,58,59]. Evidently, despite promising benefits, multiple issues need to be addressed before widescale gNBS can be integrated into health systems, emphasising the need for more research to fill these knowledge gaps. This includes an evaluation of its clinical utility and areas where unintended negative consequences may arise for affected individuals and families, in addition to exploring appropriate consent procedures for an expanded screening test, and possible storage and reinterrogation of resulting data. A key aspect of the consideration of gNBS is that it will occur within, or closely alongside, an existing and highly trusted population screening program. To this end, public acceptance and engagement with gNBS are important considerations within health system readiness. Structural and other barriers will need to be identified and addressed as research progresses[60-65].
gNBS implementation may be guided by a federated approach to the co-development of sequencing techniques that balance high sensitivity and specificity of conditions, genes, and variants against low false positive rates[66]. The development of nationally consistent approaches to consent, timing, variant curation, security of data storage and privacy is expected to facilitate effective clinical translation. Planning may safeguard against harm from screening, such as placing families into situations of ongoing uncertainty or false positive findings.
Clinical impact, consent and condition selection
The potential clinical benefits of gNBS are recognised internationally, with evidence emerging regarding its ability to facilitate earlier diagnosis of conditions that have no biochemical profile (and thus cannot be detected by tMS)[52]. Part of the evidence base for gNBS comes from existing initiatives that are evaluating the use of genomic sequencing in unwell newborns and in newborns with delayed onset or milder phenotypes[67]. Balanced against this, gNBS methodology may not be superior in terms of sensitivity or specificity for conditions already identified by tMS and included in NBS programs. Additionally, without careful variant curation (inclusive of variant analysis within Indigenous populations and ethnic subgroups[68]), it will also yield a greater number of VUS and UFs. This suggests that a complementary approach to the incorporation of genomics in NBS may be warranted instead of an “either/or” approach[69-71]. Increasing the complexity of evaluating clinical utility, the paucity of Australia-specific prevalence data for many rare conditions makes it difficult to ascertain how many children could benefit from a diagnosis through current or advancing screening techniques[72].
These issues surrounding the model of screening with emerging new technologies will also necessitate appropriate consent procedures, including a move toward an “opt-in” model of gNBS (in addition to current routine panels based on tMS). Here, it is notable that parents do not always uptake offers of extensive genetic information. One study of parents of children with congenital hearing loss receiving clinical exome sequencing revealed one-third of parents elected not to receive additional genetic information beyond their clinical results. Interestingly, parents of infants younger than three months were the least likely to request additional findings, and a significant proportion changed their decision regarding information disclosure over the study period[73]. This lower uptake by parents of young infants has been replicated across other studies and poses a potential and substantial barrier when using gNBS to expand current NBS programs[74].
Alternative models for the delivery of genetic information, such as using gNBS data as a “lifetime repository” to be accessed at relevant time points in an individual’s life, merit consideration. However, with such an approach, we must also consider the ongoing relevance and consistency in the interpretation of genetic variants and population health literacy and the ability to allocate resources for the management of this “lifetime resource”[58,65]. The cost and infrastructure required for routine storage of newborn data also need to be considered.
While there is intra- and interjurisdictional debate on which conditions are amenable, useful, and feasible for gNBS, Australian stakeholders are striving to develop pathways in which conditions can be efficiently considered for incorporation into routine panels, based on the merits of screening for each condition. One existing mechanism to promote the utility of genomic sequencing is PanelApp, deployed by Australian Genomics in 2019. This crowdsourced, publicly available knowledge-generating platform was developed to facilitate the sharing and evaluation of gene panels, contributing toward national and international efforts to establish standards of gene selection and consensus on genotype-phenotype relationships[75]. Further efforts to curate appropriate gene panels for NBS include Baby Screen +, with the proposed research criteria for condition selection including analytical and clinical validity and clinical utility (disease onset exclusively or predominantly in childhood (before five years of age), disease severity, and diseases with an effective treatment available that alters the natural history of the disease). Nongenetic functional assays are considered desirable, serving to confirm the expected phenotype and probability of symptom manifestation and reduce uncertainties associated with genes of incomplete penetrance[76]. Knowledge from international studies and other screening strategies such as RGCS[57] will also inform gene selection curation, noting that the suitability for inclusion may vary between program objectives.
Capacity building for a potential new model of newborn bloodspot screening
Capacity building will be integral to shaping a health system that is able to sustainably offer and manage increased genomic information arising from potential new models of NBS. This will include but is not limited to follow-up clinical and genetic services, public and family engagement and education to optimise genomic health literacy. Although paediatric tertiary services are situated in over half of Australian states, areas such as Tasmania and the Northern Territory rely on interstate mutual healthcare agreements for the management of children who require access to specialist genetic and clinical services. Thus, ratifying and strengthening referral pathways is essential so that no child falls through the gap of an evolving NBS program. This should include access to specialist services and coordinated management in the community.
Underpinning these needs is the imperative to engage stakeholders from genomics, NBS, rare diseases, health system, and health policy, as well as the broader community. Links between these groups should be embedded both within and between states, nationally and internationally, so that Australia can move in line with the international pace of change, embed new technologies of (newborn) screening in a health system that is ready to receive it and can overcome the obstacles that are common globally while acknowledging the uniqueness of Australia’s health system.
Potential risks and enablers of maintaining public trust
The erosion of existing high public trust in NBS is a potential risk to its expansion through the addition of conditions (either through standard or novel technologies) and should be actively considered in service planning and implementation. One such risk to be mitigated is the receipt of uncertain or unactionable information following NBS. Some studies show a high tolerance of uncertainty, for example, in a study of exome sequencing in babies with congenital deafness, where parents reported low levels of decisional regret in participating in NBS for conditions with childhood onset[73]. However, studies of population screening found that receiving uncertain results in NBS is often experienced in the same way as receiving a full genetic diagnosis and that there were significant emotional and behavioural sequelae for families[77]. Further, parents of children who receive uncertain results for conditions such as cystic fibrosis (CF) find themselves as “genetic nomads” - identifying with neither the “CF world” nor with parents of healthy children[78]. These potential risks of gNBS need to be planned for, and care delivery should include communication and adaptation strategies for receipt of uncertain results. Ensuring access to ongoing research, care and surveillance in gNBS will also be imperative if its benefits to newborns are to be realised.
High public trust in these programs will be facilitated with governance around data storage to maintain data confidentiality and integrity. Future forms of NBS will require a robust data management strategy to ensure the privacy of generated data, particularly as studies have identified concerns around the safe storage of screening results[16,79].
Public engagement will also require consideration and the implementation of measures aimed at safeguarding against potential discrimination following early genetic diagnoses facilitated by expanded NBS. Early genetic diagnoses may impact eligibility or premium prices for personal risk-rated insurance products, such as income protection and life insurance, which typically rely on an individual’s specific risk factors. There is currently an industry moratorium in place in Australia which limits the use of genetic information when determining individual risk[80]. It remains uncertain as to whether and how expanding NBS will impact the insurance discrimination landscape, although this concern (especially in relation to expanding NBS, or adding specific new conditions) has been previously raised in other jurisdictions[81,82]. Policymakers are encouraged to monitor the possible impact of expanded NBS on access to personal risk-rated products in Australia and to ensure clear communication with families.
Finally, cultural context needs to be actively planned in any expansion of NBS. Cultural background is known to shape decisions regarding whether parents seek additional genomic information in the context of clinical care[73,83], and this may also be reflected in NBS. If gNBS is to be offered to the highly heterogeneous population of Australia, there must be a balance between providing culturally sensitive, individualized support to enable effective decision making about NBS and the need to provide a standard test offered at population scale[73]. Further research is also warranted to understand the potential for stigmatisation, which in part has led to the poor uptake of NBS for haemoglobinopathies in specific ethnic groups[84].
Policy consideration for Australian newborn bloodspot screening programs: the continuing evolution of assessment methods and processes
The approach used by Australia to assess conditions for inclusion in (and potentially removal from) NBS programs continues to evolve [Figure 3]. Recently, a formal step of health technology assessment (HTA) has been added to the pathway for considering new conditions, with a requirement for assessment by the Medical Services Advisory Committee (MSAC). This is an independent non-statutory committee to provide recommendations on public reimbursement of technologies and services other than pharmaceuticals[85]. MSAC has well-established approaches for assessing the value of screening and diagnostic tests, which rely on internationally accepted methods for determining safety, effectiveness, cost-effectiveness, and budgetary impact, as well as the ethical, legal, and social implications of a technology[86].
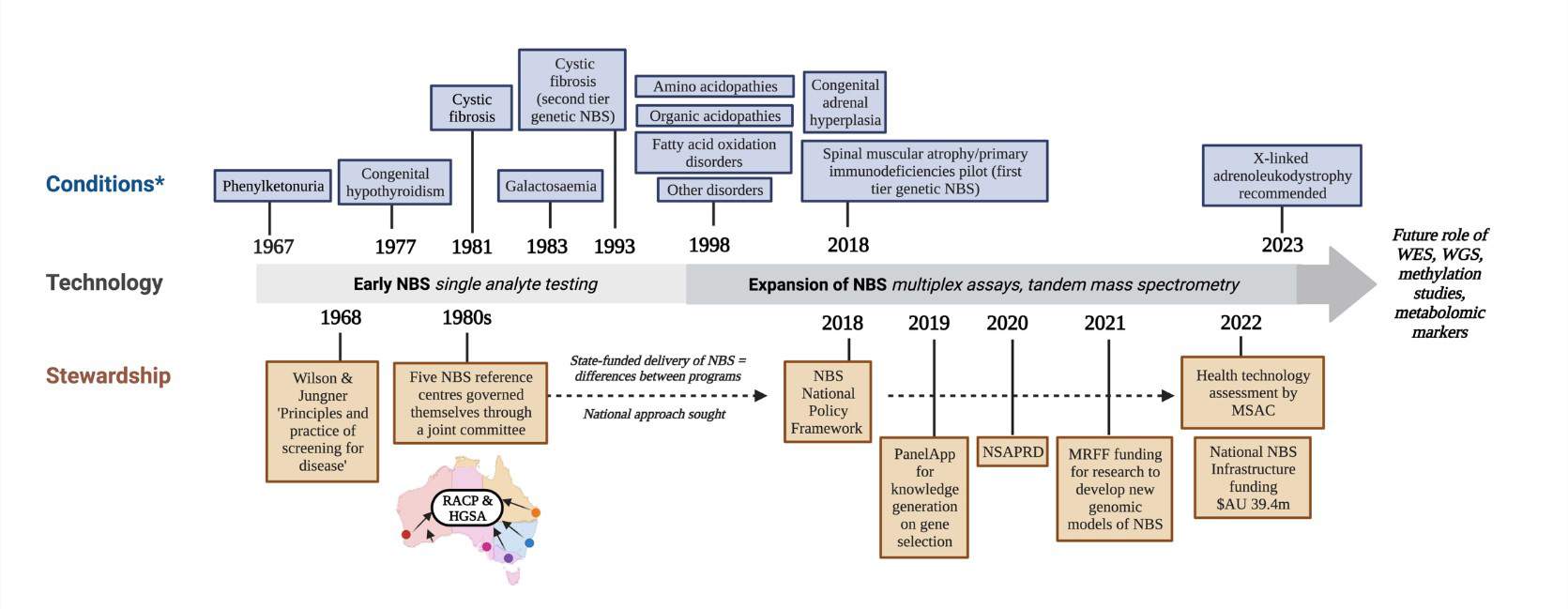
Figure 3. Fifty-five years of Australian newborn bloodspot screening[2]. This figure shows the evolution of technology and conditions screened within Australian NBS programs and key governance milestones pertaining to the stewardship of NBS programs. *Timing of the introduction of conditions screened varied between states and territories of Australia; the earliest date is indicated. RACP: Royal Australasian College of Physicians; HGSA: Human Genetics Society of Australasia; NBS: newborn bloodspot screening; NSAPRD: National Strategic Action Plan for Rare Diseases; MRFF: Medical Research Future Fund; HTA: health technology assessment; MSAC: Medical Services Advisory Committee; WES: whole exome sequencing; WGS: whole genome sequencing.
However, applying “standard” HTA methods in the context of NBS and in screening generally can be challenging. More specifically, this is due to the rarity of screened conditions (and associated issues with generating sufficient evidence), the complexity of testing and care, the broader impacts on family members, and the specific ethical, legal and social issues (ELSI) associated with the use of genetics or genomics to screen otherwise apparently healthy newborns[87,88]. In Australia, there are substantial data gaps pertaining to, for example, stakeholder perspectives regarding consent models, whether to report carrier status and/or adult-onset conditions, how to manage incidental/unsolicited findings, if/when to include conditions with incomplete penetrance or variable expressivity, and whether genomics should be used as a first-line test[25,58,59].
Harnessing the opportunities offered by advances in precision medicine and gNBS of a vast range of rare conditions requires substantial policy change with the incorporation of different approaches to generate evidence-based funding decisions[31]. Considerations include whether investment aligns with value-based health care and is sustainable, cost-effective, and socially and ethically acceptable to stakeholders.
Assessing cost-effectiveness is critical for a predominantly publicly funded screening model. This will require a comprehensive understanding of the downstream consequences for screened newborns. Potential adverse effects and expenses related to the NBS program, including novel testing modalities, follow-up care, and psychosocial impacts, must be counterbalanced against test performance and effectiveness of subsequent care. Indeed, scholars globally have called for a proportionate and nuanced approach to the introduction of genomics in NBS[60,89].
In Australia, any assessment of treatments for public reimbursement is undertaken by MSAC, the Pharmaceutical Benefits Advisory Committee (PBAC), or the Life Saving Drugs Program Expert Panel (LSDPEP). There is active coordination of public funding decisions by MSAC and PBAC for tests and treatments, with dedicated protocols for assessing “co-dependent” technologies, recognising local expectations that conditions with a publicly funded treatment should have a publicly funded test[90]. However, it is unclear how funding decisions for treatments for conditions being considered for NBS will be coordinated with decisions to screen for those conditions in the first place. Moreover, positive recommendations for public funding from one of these committees may not translate to universal access to a treatment due to the very high cost of many innovative treatments for rare conditions. Further, in Australia, a person’s eligibility to receive a public subsidy for a specific treatment may be restricted to only include a specific genotype or phenotype. In a practical sense, a person’s access may also be restricted or impacted because a treatment’s delivery requires access to a highly specialised clinical centre. Decisions about which conditions to screen for in NBS should be cognisant of this wider funding landscape. An integrated approach to NBS and research therapeutics has been recommended by a recent parliamentary enquiry[31].
Another Australian policy consideration is that the evolution of the assessment pathways for publicly funded NBS programs is occurring contemporaneously with the possible public funding of RGCS. An application for public funding for RGCS was unsuccessful[91,92], but efforts to secure public funding for expanded carrier screening are ongoing. Coordination may also be required regarding which conditions and variants are included in which program.
Ethical, legal and social issues associated with the use of genomics in newborn screening
Appropriately incorporating gNBS raises complex and nuanced questions regarding its ELSI. These arise against a backdrop of existing ELSI questions in NBS, including avoiding overdiagnosis, defining the benefits of NBS (including whether reproductive choice is a justified benefit), the scope of conditions it is appropriate to screen for, whether and how to obtain explicit consent to NBS, and ongoing access to samples.
ELSI in gNBS is the subject of a rapidly growing literature base[58,60,61,64,65,93-98]. Several of the issues discussed in this paper have an ethical dimension, too: consent, managing uncertainty, whether to disclose incidental or unsolicited findings, choosing which conditions to screen, disclosing carrier status, and the possibility of discrimination. There are additional factors, such as tensions between public health and clinical paradigms (and the expectations and limitations these give rise to), as well as issues such as what should constitute a “benefit” of gNBS[99]. Questions of benefit in gNBS require particular attention, such as whether secondary benefits to the family justify reporting certain results even when the newborn themselves may not benefit, or whether to report variants for conditions with X-linked inheritance when these are detected in female newborns. A further aspect of “benefit” is whether to screen for a condition when only a proportion of screen-positive newborns (for instance, only those with a particular genotype) will be able to access a treatment or risk-reducing intervention.
Many of the ethical issues around the use of gNBS are not related to the technology itself, but to the context in which the technology is being used: population screening of apparently healthy babies. An appropriate ethical framing in this context is public health ethics, which emphasises public goods, social justice, and community benefits. On this view, populations are more than aggregates of individuals - the socio-political context is vital to consider as well. Issues such as population-level harms (e.g., through overdiagnosis), structural barriers to health, and increasing the individualisation of responsibility for health are all relevant. Ethical concepts and values such as solidarity, trust (discussed earlier in this paper), and reciprocity are important to consider alongside more widely explored concepts such as autonomy. Attending to the nuances that an analysis of these additional concepts draws out will enable a more equitable and appropriate use of gNBS[65].
In addition to ensuring universal access to screening and treatments, other key equity aspects related to gNBS include recognition of the sovereignty of Indigenous Australians’ genomic data, increasing the representation of our diverse populations in the genetic databases used in screening[68], and developing a clear and transparent process for prioritising which conditions are considered for inclusion in NBS programs. If a condition is not prevalent in all population groups, it is important to design screening to actively mitigate against identifying and reporting false negative results.
CONCLUSION
NBS programs in Australia have been a central component of the country’s ability to effectively diagnose and facilitate effective therapeutic interventions to generations of children with often rare conditions, unifying healthcare delivery across a heterogenous and widely dispersed population. The changing environment and unmet needs of people and families living with rare conditions are driving active discourses on the principles of screening through a modern lens, serving to navigate the opportunities and challenges of genomic technologies in expanding NBS programs. These steps will support the future collaborative development of Australian NBS programs in line with innovation, being both person- and family-centred and informed by cumulative local and international knowledge and evidence. Poised on the precipice of a genomic revolution, NBS in Australia has the capability to expand health benefits to broader populations; however, a coordinated, national approach and prospective evaluation of outcomes and processes are essential so that NBS can safely, equitably, and effectively adapt for the future, evolving to meet the changing needs of the populations it serves[100].
DECLARATIONS
Authors’ contributions
Formulated the concept of the manuscript: Ji C, Farrar MA, Kariyawasam DS, Norris S, Ji C, Farrar MA.
Contributed to the first and subsequent drafts: Kariyawasam DS, Norris S, Newson AJ, Bhattacharya K, Bennetts B
Contributed to multiple critical revisions: Ji C, Farrar MA, Norris S, Bhattacharya K, Bennetts B, Newson AJ, Healy L, Millis N, Kariyawasam DS
Availability of data and materials
Not applicable.
Financial support and sponsorship
Farrar MA, Bhattacharya K and Bennetts B are recipients of grant support from the Medical Research Futures (fund GNT2017165), “newborn gen seq trail: newborn genomicsequencing in screening: therapy ready and information for life”; Norris S, Newson AJ, and Kariyawasam DS are recipients of grant support from the Medical Research Futures (fund MRF2015965), “gEnomics4newborns: integrating Ethics and Equity with Effectiveness and Economics for genomic newborn screening”; Newson AJ is the recipient of grant support from the Medical Research Futures (fund MRF2015531), “Ethical governance of clinical and genomic datasets, now known as the LINEAGE Project”.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
1. Human Genetics Society of Australasia. Counting conditions and summary of conditions screened by programme. Available from: https://hgsa.org.au/common/Uploaded%20files/pdfs/policies,%20position%20statements%20and%20guidelines/newborn%20screening/2023%20PL01%20Counting%20Conditions%20and%20Summary%20of%20Conditions%20Screened%20by%20Programme..pdf [Last accessed on 21 Nov 2023].
3. The Department of Health and Aged Care. Population-based screening framework. Available from: https://www.health.gov.au/resources/publications/population-based-screening-framework?language=en [Last accessed on 21 Nov 2023].
4. Wilson JMG, Jungner G. Principles and practice of screening for disease. Geneva: World Health Organisation;1968. Available from: https://niercheck.nl/wp-content/uploads/2019/06/Wilson-Jungner-1968.pdf [Last accessed on 21 Nov 2023].
5. Coman D, Bhattacharya K. Extended newborn screening: an update for the general paediatrician. J Paediatr Child Health 2012;48:E68-72.
6. Franková V, Driscoll RO, Jansen ME, et al; Members of the European Society of Human Genetics (ESHG)-EuroGentest Quality Sub-Committee. Regulatory landscape of providing information on newborn screening to parents across Europe. Eur J Hum Genet 2021;29:67-78.
7. Australian Health Ministers’ Advisory Council. Newborn bloodspot screening national policy framework. Available from: https://www.health.gov.au/sites/default/files/documents/2020/10/newborn-bloodspot-screening-national-policy-framework.pdf [Last accessed on 21 Nov 2023].
8. Kariyawasam D, Russell JS, Wiley V, Alexander IE, Farrar MA. The implementation of newborn screening for spinal muscular atrophy: the Australian experience. Genet Med 2020;22:557-65.
9. Kariyawasam DS, D'Silva AM, Sampaio H, et al. Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study. Lancet Child Adolesc Health 2023;7:159-70.
10. Dangouloff T, Boemer F, Servais L. Newborn screening of neuromuscular diseases. Neuromuscul Disord 2021;31:1070-80.
11. Shih STF, Keller E, Wiley V, Wong M, Farrar MA, Chambers GM. Economic evaluation of newborn screening for severe combined immunodeficiency. Int J Neonatal Screen 2022;8:44.
12. Shih STF, Keller E, Wiley V, Farrar MA, Wong M, Chambers GM. Modelling the cost-effectiveness and budget impact of a newborn screening program for spinal muscular atrophy and severe combined immunodeficiency. Int J Neonatal Screen 2022;8:45.
13. Huang X, Wu D, Zhu L, et al. Application of a next-generation sequencing (NGS) panel in newborn screening efficiently identifies inborn disorders of neonates. Orphanet J Rare Dis 2022;17:66.
14. Godler DE, Ling L, Gamage D, et al. Feasibility of screening for chromosome 15 imprinting disorders in 16 579 newborns by using a novel genomic workflow. JAMA Netw Open 2022;5:e2141911.
15. Bourmaud A, Gallien S, Domon B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: principle and applications. Proteomics 2016;16:2146-59.
16. Holm IA, Agrawal PB, Ceyhan-Birsoy O, et al; BabySeq Project Team. The babySeq project: implementing genomic sequencing in newborns. BMC Pediatr 2018;18:225.
17. Roman TS, Crowley SB, Roche MI, et al. Genomic sequencing for newborn screening: results of the NC NEXUS project. Am J Hum Genet 2020;107:596-611.
18. Ceyhan-Birsoy O, Murry JB, Machini K, et al; BabySeq Project Team. Interpretation of genomic sequencing results in healthy and ill newborns: results from the babyseq project. Am J Hum Genet 2019;104:76-93.
19. Sagan A, McDaid D, Rajan S, Farrington J, McKee M. Screening: when is it appropriate and how can we get it right? Copenhagen (Denmark): European Observatory on Health Systems and Policies; 2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559794/ [Last accessed on 21 Nov 2023].
20. Andermann A, Blancquaert I, Beauchamp S, Déry V. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ 2008;86:317-9.
21. Andermann A, Blancquaert I, Beauchamp S, Costea I. Guiding policy decisions for genetic screening: developing a systematic and transparent approach. Public Health Genomics 2010;14:9-16.
22. Andermann A, Blancquaert I, Déry V. Genetic screening: a conceptual framework for programmes and policy-making. J Health Serv Res Policy 2010;15:90-7.
23. Arah OA, Klazinga NS, Delnoij DM, ten Asbroek AH, Custers T. Conceptual frameworks for health systems performance: a quest for effectiveness, quality, and improvement. Int J Qual Health Care 2003;15:377-98.
24. Australian Institute of Health and Welfare. Health expenditure Australia 2020-21. Available from: https://www.aihw.gov.au/reports/health-welfare-expenditure/health-expenditure-australia-2020-21/contents/spending-trends-by-sources [Last accessed on 21 Nov 2023].
25. Cao M, Notini L, Ayres S, Vears DF. Australian healthcare professionals' perspectives on the ethical and practical issues associated with genomic newborn screening. J Genet Couns 2023;32:376-86.
26. Metz MP, Ranieri E, Gerace RL, Priest KR, Luke CG, Chan A. Newborn screening in South Australia: is it universal? Med J Aust 2003;179:412-5.
27. Nolan-Isles D, Macniven R, Hunter K, et al. Enablers and barriers to accessing healthcare services for aboriginal people in New South Wales, Australia. Int J Environ Res Public Health 2021;18:3014.
28. D'Silva AM, Kariyawasam DST, Best S, Wiley V, Farrar MA; NSW SMA NBS Study Group. Integrating newborn screening for spinal muscular atrophy into health care systems: an Australian pilot programme. Dev Med Child Neurol 2022;64:625-32.
29. Therrell BL, Padilla CD, Loeber JG, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol 2015;39:171-87.
30. Department of Health and Aged Care. Expansion of newborn bloodspot screening. Available from: https://www.health.gov.au/our-work/newborn-bloodspot-screening/expansion [Last accessed on 21 Nov 2023].
31. Commonwealth of Australia. The new frontier - delivering better health for all Australians. Available from: https://www.aph.gov.au/Parliamentary_Business/Committees/House/Health_Aged_Care_and_Sport/Newdrugs/Report [Last accessed on 21 Nov 2023].
32. Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med 2003;348:2304-12.
33. Hall PL, Marquardt G, Mchugh DM, et al. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med 2014;16:889-95.
34. Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, Rodney Howell R. Newborn screening: toward a uniform screening panel and system. Genet Med 2006;8 Suppl 1:1S-252S.
35. Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 2003;111:1399-406.
36. Forny P, Hörster F, Ballhausen D, et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: first revision. J Inherit Metab Dis 2021;44:566-92.
37. Hertzog A, Selvanathan A, Pandithan D, et al. 3-Methylglutaconyl-CoA hydratase deficiency: when ascertainment bias confounds a biochemical diagnosis. JIMD Rep 2022;63:568-74.
38. Thompson S, Hertzog A, Selvanathan A, et al. Treatment of HMG-CoA lyase deficiency-longitudinal data on clinical and nutritional management of 10 australian cases. Nutrients 2023;15:531.
39. Rüfenacht V, Häberle J. Mini-review: challenges in newborn screening for urea cycle disorders. Int J Neonatal Screen 2015;1:27-35.
40. California Department of Public Health. NEwborn screening program: disorders detectable by newborn screening. Available from: https://www.cdph.ca.gov/Programs/CFH/DGDS/Pages/nbs/NBS-Disorders-Detectable.aspx [Last accessed on 21 Nov 2023].
41. Ryder B, Inbar-Feigenberg M, Glamuzina E, et al. New insights into carnitine-acylcarnitine translocase deficiency from 23 cases: management challenges and potential therapeutic approaches. J Inherit Metab Dis 2021;44:903-15.
42. Health Resources & Services Administration. Previously nominated conditions. Available from: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/previous-nominations [Last accessed on 21 Nov 2023].
43. Department of Health and Aged Care. What is screened in the program. Available from: https://www.health.gov.au/our-work/newborn-bloodspot-screening/what-is-screened [Last accessed on 21 Nov 2023].
44. Department of Health and Aged Care. Delivering newborn bloodspot screening programs. Available from: https://www.health.gov.au/our-work/newborn-bloodspot-screening/delivering-programs [Last accessed on 21 Nov 2023].
45. Farrar MA, Kiernan MC. Spinal muscular atrophy - the dawning of a new era. Nat Rev Neurol 2020;16:593-4.
46. McPherson E. Postcode lottery: The screening test saving babies in some states but not others. Available from: https://www.9news.com.au/national/newborn-screening-australia-postcode-lottery-calls-for-heel-prick-test-to-be-expanded/39cad501-9109-4855-8c59-e30c700903a1 [Last accessed on 21 Nov 2023].
47. Newborn screening: a blueprint for the future executive summary: newborn screening task force report. Pediatrics 2000;106:386-8.
48. Bhattacharya K, Millis N, Jaffe A, Zurynski Y. Rare diseases research and policy in Australia: On the journey to equitable care. J Paediatrics Child Health 2021;57:778-81.
49. Australian Government Department of Health and Aged Care. National strategic action plan for rare diseases. Available from: https://www.health.gov.au/resources/publications/national-strategic-action-plan-for-rare-diseases [Last accessed on 21 Nov 2023].
50. Department of Health and Aged Care. MRFF genomics health futures mission implementation plan. Available from: https://www.health.gov.au/resources/publications/mrff-genomics-health-futures-mission-implementation-plan?language=en [Last accessed on 21 Nov 2023].
51. Australian Genomics. Genomic screening consortium for australian newborns (GenSCAN). Available from: https://www.australiangenomics.org.au/projects/genomic-screening-consortium-for-australian-newborns-genscan/ [Last accessed on 21 Nov 2023].
52. Kingsmore SF, Smith LD, Kunard CM, et al. A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. Am J Hum Genet 2022;109:1605-19.
53. Owen MJ, Lefebvre S, Hansen C, et al. An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases. Nat Commun 2022;13:4057.
54. Nurchis MC, Altamura G, Riccardi MT, et al. Whole genome sequencing diagnostic yield for paediatric patients with suspected genetic disorders: systematic review, meta-analysis, and GRADE assessment. Arch Public Health 2023;81:93.
55. Sachdev R, Field M, Baynam GS, et al. Paediatric genomic testing: navigating medicare rebatable genomic testing. J Paediatr Child Health 2021;57:477-83.
56. Burdick KJ, Cogan JD, Rives LC, et al; Undiagnosed Diseases Network. Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am J Med Genet A 2020;182:1400-6.
57. Kirk EP, Ong R, Boggs K, et al. Gene selection for the Australian reproductive genetic carrier screening project (“Mackenzie's Mission”). Eur J Hum Genet 2021;29:79-87.
58. Vears DF, Savulescu J, Christodoulou J, Wall M, Newson AJ. Are we ready for whole population genomic sequencing of asymptomatic newborns? Pharmgenomics Pers Med 2023;16:681-91.
59. White S, Mossfield T, Fleming J, et al. Expanding the Australian newborn blood spot screening program using genomic sequencing: do we want it and are we ready? Eur J Hum Genet 2023;31:703-11.
60. Johnston J, Lantos JD, Goldenberg A, Chen F, Parens E, Koenig BA; members of the NSIGHT Ethics and Policy Advisory Board. Sequencing newborns: a call for nuanced use of genomic technologies. Hastings Center Report 2018; 48 Suppl 2:S2-6.
61. Ross LF, Clayton EW. Ethical issues in newborn sequencing research: the case study of babyseq. Pediatrics 2019;144:e20191031.
63. Botkin JR, Rothwell E. Whole genome sequencing and newborn screening. Curr Genet Med Rep 2016;4:1-6.
64. Woerner AC, Gallagher RC, Vockley J, Adhikari AN. The use of whole genome and exome sequencing for newborn screening: challenges and opportunities for population health. Front Pediatr 2021;9:663752.
65. Newson AJ. The promise of public health ethics for precision medicine: the case of newborn preventive genomic sequencing. Hum Genet 2022;141:1035-43.
66. Pichini A, Ahmed A, Patch C, et al. Developing a national newborn genomes program: an approach driven by ethics, engagement and co-design. Front Genet 2022;13:866168.
67. Bhattacharjee A, Sokolsky T, Wyman SK, et al. Development of DNA confirmatory and high-risk diagnostic testing for newborns using targeted next-generation DNA sequencing. Genet Med 2015;17:337-47.
68. Easteal S, Arkell RM, Balboa RF, et al; National Centre for Indigenous Genomics. Equitable expanded carrier screening needs indigenous clinical and population genomic data. Am J Hum Genet 2020;107:175-82.
69. Ko JM, Park KS, Kang Y, et al. A new integrated newborn screening workflow can provide a shortcut to differential diagnosis and confirmation of inherited metabolic diseases. Yonsei Med J 2018;59:652-61.
70. Wojcik MH, Zhang T, Ceyhan-Birsoy O, et al; BabySeq Project Team. Discordant results between conventional newborn screening and genomic sequencing in the babyseq project. Genet Med 2021;23:1372-5.
71. Downie L, Halliday J, Lewis S, Amor DJ. Principles of genomic newborn screening programs: a systematic review. JAMA Netw Open 2021;4:e2114336.
72. Walker CE, Mahede T, Davis G, et al. The collective impact of rare diseases in Western Australia: an estimate using a population-based cohort. Genetics in Medicine 2017;19:546-52.
73. Downie L, Halliday J, Lewis S, et al. Exome sequencing in newborns with congenital deafness as a model for genomic newborn screening: the baby beyond hearing project. Genet Med 2020;22:937-44.
74. Genetti CA, Schwartz TS, Robinson JO, et al; BabySeq Project Team. Parental interest in genomic sequencing of newborns: enrollment experience from the babyseq project. Genet Med 2019;21:622-30.
75. Martin AR, Williams E, Foulger RE, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet 2019;51:1560-5.
76. Yeung A, Christodoulou J, Amor D, et al. Baby Screen+ newborn screening (version 1.108). Available from: https://panelapp.agha.umccr.org/panels/3931/ [Last accessed on 21 Nov 2023].
77. Johnson F, Ulph F, MacLeod R, Southern KW. Receiving results of uncertain clinical relevance from population genetic screening: systematic review & meta-synthesis of qualitative research. Eur J Hum Genet 2022;30:520-31.
78. Boardman F, Clark C. 'We're kind of like genetic nomads': prents' experiences of biographical disruption and uncertainty following in/conclusive results from newborn cystic fibrosis screening. Soc Sci Med 2022;301:114972.
79. Esquerda M, Palau F, Lorenzo D, et al. Ethical questions concerning newborn genetic screening. Clin Genet 2021;99:93-8.
80. Financial Services Council. FSC Standard No. 11: moratorium on genetic tests in life insurance. Available from: www.fsc.org.au/resources-category/standard/1779-standard-11-moratorium-on-genetic-tests-in-life-insurance/file [Last accessed on 21 Nov 2023].
81. Dhondt JL. Expanded newborn screening: social and ethical issues. J Inherit Metab Dis 2010;33:S211-7.
82. Zacharias RL, Smith ME, King JS. The legal dimensions of genomic sequencing in newborn screening. Hastings Cent Rep 2018;48 Suppl 2:S39-41.
83. Lakes KD, Vaughan E, Lemke A, et al. Maternal perspectives on the return of genetic results: context matters. Am J Med Genet A 2013;161A:38-47.
84. Verma IC, Saxena R, Kohli S. Past, present & future scenario of thalassaemic care & control in India. Indian J Med Res 2011;134:507-21.
85. Medical Services Advisory Committee. About MSAC. Available from: http://msac.gov.au/internet/msac/publishing.nsf/Content/about-msac [Last accessed on 21 Nov 2023].
86. Medical Services Advisory Committee. Guidelines for preparing assessments for the Medical Services Advisory Committee. Available from: http://www.msac.gov.au/internet/msac/publishing.nsf/Content/E0D4E4EDDE91EAC8CA2586E0007AFC75/$File/MSAC%20Guidelines-complete-16-FINAL(18May21).pdf [Last accessed on 21 Nov 2023].
87. Potter BK, Avard D, Entwistle V, et al. Ethical, legal, and social issues in health technology assessment for prenatal/preconceptional and newborn screening: a workshop report. Public Health Genomics 2009;12:4-10.
88. Potter BK, Avard D, Graham ID, et al. Guidance for considering ethical, legal, and social issues in health technology assessment: application to genetic screening. Int J Technol Assess Health Care 2008;24:412-22.
89. Rahimzadeh V, Friedman JM, de Wert G, Knoppers BM. Exome/genome-wide testing in newborn screening: a proportionate path forward. Front Genet 2022;13:865400.
90. Pharmaceutical Benefits Advisory Committee. About the guidelines. Available from: https://pbac.pbs.gov.au/information/about-the-guidelines.html [Last accessed on 21 Nov 2023].
91. Medical Services Advisory Committee. Application No. 1573 - reproductive carrier testing for cystic fibrosis, spinal muscular atrophy and fragile X syndrome. Available from: http://www.msac.gov.au/internet/msac/publishing.nsf/Content/4EF0E3C5A7CC9D05CA2584240009557E/$File/1573%20-%20Final%20PSD_Jul2020.pdf [Last accessed on 21 Nov 2023].
92. Medical Services Advisory Committee. Application No. 1637 - expanded reproductive carrier testing of couples for joint carrier status of genes associated with autosomal recessive and X-linked conditions. Available from: http://www.msac.gov.au/internet/msac/publishing.nsf/Content/58B9ED94DEC5BCDECA2586D500054D7E/$File/1637%20Final%20PSD_Jul2022_redacted_UpdatedConsultation_clean.pdf [Last accessed on 21 Nov 2023].
93. Ulph F, Bennett R. Psychological and ethical challenges of introducing whole genome sequencing into routine newborn screening: lessons learned from existing newborn screening. New Bioeth 2023;29:52-74.
94. Williams WA 2nd, Ross LF. The Harms of Carrier Status Identification: a cautionary warning against newborn sequencing. J Pediatr 2020;224:22-3.
95. Goldenberg AJ, Lloyd-Puryear M, Brosco JP, et al; Bioethics and Legal Workgroup of the Newborn Screening Translational Research Network. Including ELSI research questions in newborn screening pilot studies. Genet Med 2019;21:525-33.
96. Currier RJ. Newborn screening is on a collision course with public health ethics. Int J Neonatal Screen 2022;8:51.
97. Kemper AR, Boyle CA, Brosco JP, Grosse SD. Ensuring the life-span benefits of newborn screening. Pediatrics 2019;144:e20190904.
98. Sen K, Harmon J, Gropman AL. Select ethical aspects of next-generation sequencing tests for newborn screening and diagnostic evaluation of critically Ill newborns. Int J Neonatal Screen 2021;7:76.
99. Clark CC, Boardman FK. Expanding the notion of “benefit”: comparing public, parent, and professional attitudes towards whole genome sequencing in newborns. New Genetics and Society 2022;41:96-115.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ji C, Farrar MA, Norris S, Bhattacharya K, Bennetts B, Newson AJ, Healy L, Millis N, Kariyawasam DS. The Australian landscape of newborn screening in the genomics era. Rare Dis Orphan Drugs J 2023;2:26. http://dx.doi.org/10.20517/rdodj.2023.30
AMA Style
Ji C, Farrar MA, Norris S, Bhattacharya K, Bennetts B, Newson AJ, Healy L, Millis N, Kariyawasam DS. The Australian landscape of newborn screening in the genomics era. Rare Disease and Orphan Drugs Journal. 2023; 2(4): 26. http://dx.doi.org/10.20517/rdodj.2023.30
Chicago/Turabian Style
Ji, Charli, Michelle A Farrar, Sarah Norris, Kaustuv Bhattacharya, Bruce Bennetts, Ainsley J Newson, Louise Healy, Nicole Millis, Didu S Kariyawasam. 2023. "The Australian landscape of newborn screening in the genomics era" Rare Disease and Orphan Drugs Journal. 2, no.4: 26. http://dx.doi.org/10.20517/rdodj.2023.30
ACS Style
Ji, C.; Farrar MA.; Norris S.; Bhattacharya K.; Bennetts B.; Newson AJ.; Healy L.; Millis N.; Kariyawasam DS. The Australian landscape of newborn screening in the genomics era. Rare. Dis. Orphan. Drugs. J. 2023, 2, 26. http://dx.doi.org/10.20517/rdodj.2023.30
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 3
likes
Like This Article 3
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.